Multiple system atrophy
Peer reviewed by Dr Laurence KnottLast updated by Dr Colin Tidy, MRCGPLast updated 16 Mar 2021
Meets Patient’s editorial guidelines
Medical Professionals
Professional Reference articles are designed for health professionals to use. They are written by UK doctors and based on research evidence, UK and European Guidelines. You may find one of our health articles more useful.
In this article:
Continue reading below
Overview
Multiple system atrophy (MSA) is a rare neurodegenerative disorder, caused by cell loss in certain areas of the brain and the spinal cord, leading to a variety of symptoms affecting especially the functions of the autonomic nervous system and the motor system. These are characterised by Parkinsonian features of varying severity, cerebellar ataxia and autonomic (particularly urogenital) dysfunction1. There may also be some corticospinal features.
The aetiology is not fully understood2. Novel treatment options are being investigated but currently management options are very limited. There is no cure.
MSA is referred to as MSA-P type if Parkinsonian features predominate3. The terms striatonigral degeneration or Parkinsonian variant are sometimes used in these cases.
MSA-C type describes disease where cerebellar symptoms predominate3. This may also be described as sporadic olivopontocerebellar atrophy.
The term Shy-Drager syndrome, which was used to describe MSA with predominant autonomic dysfunction, is not now used, as almost every patient is affected by autonomic or urinary dysfunction.
Aetiology4
MSA is characterised by widespread glial cytoplasmic inclusions (GCIs) which are the hallmark of the disease. More recently, misfolded, hyperphosphorylated fibrillar α-synuclein has been identified as the main component of GCIs5.
The density of GCI containing α-synuclein correlates significantly with neuronal deterioration and disease duration. Another important protein, p25α has been found to stimulate α-synuclein in vitro. It is thought that there may be both genetic and environmental processes that contribute to these pathological processes.
The presence of GCIs is associated with neuronal loss in the basal ganglia, cerebellum, pons, inferior olivary nuclei and the spinal cord, hence giving rise to the spectrum of symptoms and clinical findings. Disease is often defined at the time of initial manifestation of any motor or autonomic features, although subclinical neuropathology is likely to start several years before overt disease.
Continue reading below
Epidemiology
MSA is a progressive neurodegenerative disorder with an estimated annual worldwide incidence of about 0·6/100,000, rising to 3/100,000 in those aged over 50 years6.
Some say that there is no gender difference4. However, others report a higher diagnosis rate in males, with a ratio of male:female of between 1:3 and 1:93.
Most patients with MSA develop the disease when older than 40 years and the average age of onset is approximately 55 years7.
Presentation
The first symptoms are often autonomic and may predate recognition of motor manifestations. Orthostatic hypotension and, in men, erectile failure are among the first symptoms7.
Patients may also present with Parkinsonian symptoms, often with a poor or temporary response to levodopa therapy, or cerebellar dysfunction.
Corticospinal tract dysfunction may occur but is not usually a major presentation.
When the disorder presents with non-autonomic features, imbalance caused by cerebellar or extrapyramidal abnormalities is the most common feature.
Constipation may also occur.
There may possibly be mild intellectual impairment8. This is particularly so in older patients with greater physical disability9.
Other neuropsychiatric problems may include depression, insomnia, daytime sleepiness, restless legs, hallucinations and dementia4.
Continue reading below
Diagnosis
The diagnosis of MSA is based mainly on clinical features. Most patients do not receive the correct diagnosis during their lifetime because of the difficulty in differentiation from other disorders, particularly Parkinson's disease and pure autonomic failure.
Definite diagnosis can only be made post-mortem. However, major and additional features have been identified which support a possible diagnosis. Additionally, there are some aspects of the history and examination that go against the diagnosis.
Thus levels of certainty of diagnosis are described, depending on the clinical findings and investigations. Categories include definitive MSA, probable MSA and possible MSA2.
Major features supporting diagnosis of probable MSA
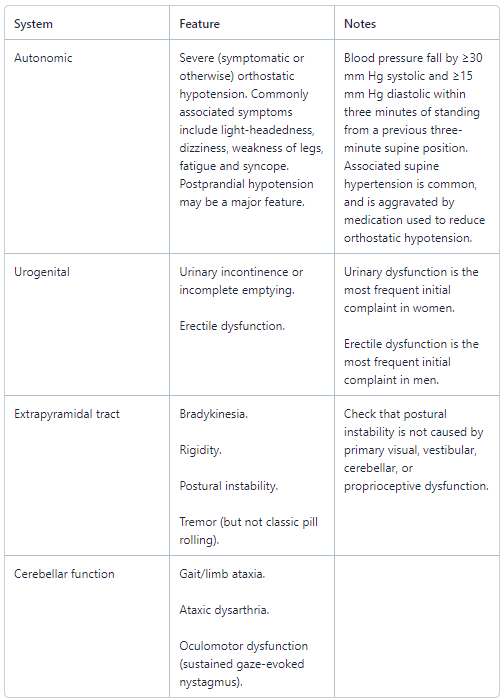
Additional features supporting diagnosis of possible MSA4
Sporadic, progressive disease of onset after 30 years of age, characterised by:
Parkinsonism.
Cerebellar signs.
At least one feature suggesting autonomic dysfunction (eg, urinary symptoms, erectile dysfunction, orthostatic hypotension that doesn't meet the level required in 'probable MSA' - see table above).
At least one of the features in the table below:
MSA type | Feature |
MSA-P or MSA-C | Babinski's sign with hyperreflexia. Stridor. |
MSA-P | Rapidly progressive Parkinsonism with poor response to levodopa. Postural instability within three years of motor onset. Gait ataxia, cerebellar dysarthria, limb ataxia or cerebellar oculomotor dysfunction. Dysphagia within five years of motor onset. |
MSA-C | Parkinsonism (bradykinesia and rigidity). Atrophy on MRI of putamen, middle cerebellar peduncle, or pons. |
Features suggesting alternative diagnosis
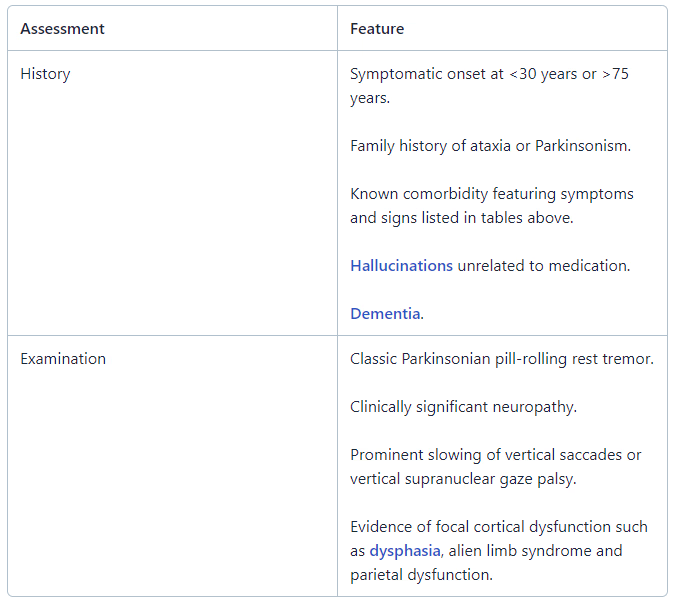
Differential diagnosis
Parkinson's disease is the main differential; about 10% of patients diagnosed with Parkinson's disease are actually found to have MSA on autopsy. Features that suggest MSA over Parkinson's disease include:
Rapid progression of symptoms.
Poor response to levodopa.
More pronounced autonomic features.
Rigidity and bradykinesia being out of proportion to the tremor.
Speech which may be severely affected.
Aspiration, inspiratory gasps and stridor which may be present.
Other diagnoses to consider include:
Pure autonomic failure.
Progressive supranuclear palsy (Steele-Richardson-Olszewski disease).
Neuroacanthocytosis.
Neurosarcoidosis.
Neurosyphilis.
Investigations
Diagnostic techniques include structural and functional brain imaging, cardiac sympathetic imaging, cardiovascular autonomic testing, olfactory testing, sleep study, urological evaluation, and dysphagia and cognitive assessments10.
Autonomic function testing4
Bladder function assessment often detects early abnormalities consistent with neurogenic disturbance. Initially, detrusor hyperreflexia and abnormal urethral sphincter function predominate; these are later followed by increased residual urinal volume (as detected by bladder ultrasound). Other autonomic abnormalities include:
Diminished respiratory sinus arrhythmia.
Abnormal response to the Valsalva manoeuvre (no blood pressure recovery in late phase II and/or no overshoot in phase IV).
Diminished response to isometric exercise (hand grip).
Diminished response to cold pressor stimuli.
Iodine-123 (I-123) metaiodobenzylguanidine (MIBG) scintigraphy
Thought to be useful for differentiation between Parkinson's disease and MSA early after onset of autonomic dysfunction11.
Patients with Parkinson's disease have significantly lower cardiac uptake of I-123 MIBG than patients with MSA and controls.
MRI and proton magnetic resonance
Brain imaging may be normal in MSA. Localised brain degeneration may be detected by MRI techniques.
The slit hyperintensity of the lateral margin of the putamen in T2-weighted MRI is a characteristic finding in patients with MSA, involving the extrapyramidal system.
Fluoride 2-(F18)fluoro-2-deoxy-D-glucose (FDG) positron emission tomography (PET) imaging
Can be used for differentiation between MSA and Parkinson's disease.
The caudate-putamen index (difference in the uptakes in the caudate and putamen divided by the caudate uptake) is lower in patients with MSA than in patients with Parkinson's disease.
Histology
Neuropathological changes consist of a high density of glial cytoplasmic inclusions in association with degenerative changes in certain brain structures - eg, putamen, caudate nucleus, globus pallidus, thalamus, pontine nuclei, cerebellar Purkinje's cells and autonomic nuclei of the brain stem.
Glial cytoplasmic inclusions: can be stained by the Gallyas silver technique and are a hallmark of MSAs.
Management
Currently, no therapy can reverse or halt progression of the disease. Management is symptomatic and targets Parkinsonism and autonomic failure4.
The extrapyramidal and cerebellar aspects of the disease are debilitating and difficult to treat. Orthostatic hypotension is associated with reduced physical activity (and the consequent deconditioning and problems associated with this) so management of this is a particularly important aspect of patient care. Management of patients with MSA will include:
Management of postural hypotension: see the separate Hypotension article.
Management of constipation, urinary incontinence and falls.
Physical activity, especially in water, to prevent physical deconditioning.
Speech therapy which may be required to help with speech and swallowing.
Movement disorder: usually treated with levodopa, dopaminergic agonists, anticholinergic agents, or amantadine, but effectiveness may be limited.
Recombinant erythropoietin: increases the functional capacity of patients, particularly if there is associated mild anaemia, which is common. Recombinant erythropoietin has been shown to correct anaemia and improve standing blood pressure.
Other agents that are much less often used include non-steroidal anti-inflammatory drugs, antihistamines, somatostatin analogues, caffeine, and yohimbine.
Future therapeutic options4
At the moment, no neuroprotective treatment is available12. However, there are potential drug candidates that have been considered:
Growth hormone therapy: experimentally, growth hormone therapy appears to slow progression of the disease but not significantly.
Minocycline: this is a tetracycline with neuroprotective efficacy in transgenic MSA mice which has shown some promise in the early stages of the disease in laboratory studies.
Rasagiline: this is a monoamine-oxidase-B inhibitor which appears to have disease-modifying effects and is soon expected to enter phase 3 trials.
Rifampicin: this has been shown to have the property of preventing α-synuclein aggregation and so is also being considered as a therapeutic candidate.
Prognosis
The clinical symptoms are rapidly progressing with a mean life expectancy following diagnosis of seven years7.
One review found prognostic indicators of shorter survival were older age at onset, early bladder catheterisation, and early generalised autonomic failure13.
Bronchopneumonia and sudden death are common terminal events.
Further reading and references
- Ahmed Z, Asi YT, Sailer A, et al; The neuropathology, pathophysiology and genetics of multiple system atrophy. Neuropathol Appl Neurobiol. 2012 Feb;38(1):4-24. doi: 10.1111/j.1365-2990.2011.01234.x.
- Jellinger KA; Multiple System Atrophy: An Oligodendroglioneural Synucleinopathy1. J Alzheimers Dis. 2018;62(3):1141-1179. doi: 10.3233/JAD-170397.
- Bjornsdottir A, Gudmundsson G, Blondal H, et al; Incidence and prevalence of multiple system atrophy: a nationwide study in Iceland. J Neurol Neurosurg Psychiatry. 2013 Feb;84(2):136-40. doi: 10.1136/jnnp-2012-302500. Epub 2012 Nov 28.
- Stefanova N, Bucke P, Duerr S, et al; Multiple system atrophy: an update. Lancet Neurol. 2009 Dec;8(12):1172-8.
- Wenning GK, Krismer F; Multiple system atrophy. Handb Clin Neurol. 2013;117:229-41. doi: 10.1016/B978-0-444-53491-0.00019-5.
- Ubhi K, Low P, Masliah E; Multiple system atrophy: a clinical and neuropathological perspective. Trends Neurosci. 2011 Nov;34(11):581-90. doi: 10.1016/j.tins.2011.08.003. Epub 2011 Sep 29.
- McKay JH, Cheshire WP; First symptoms in multiple system atrophy. Clin Auton Res. 2018 Apr;28(2):215-221. doi: 10.1007/s10286-017-0500-0. Epub 2018 Jan 8.
- European Multiple System Atrophy Study Group
- Brown RG, Lacomblez L, Landwehrmeyer BG, et al; Cognitive impairment in patients with multiple system atrophy and progressive Multiple system atrophy: an update. Brain. 2010 Jun 24.
- Palma JA, Norcliffe-Kaufmann L, Kaufmann H; Diagnosis of multiple system atrophy. Auton Neurosci. 2018 May;211:15-25. doi: 10.1016/j.autneu.2017.10.007. Epub 2017 Oct 23.
- Treglia G, Stefanelli A, Cason E, et al; Diagnostic performance of iodine-123-metaiodobenzylguanidine scintigraphy in differential diagnosis between Parkinson's disease and multiple-system atrophy: a systematic review and a meta-analysis. Clin Neurol Neurosurg. 2011 Dec;113(10):823-9. doi: 10.1016/j.clineuro.2011.09.004. Epub 2011 Oct 2.
- Kuzdas-Wood D, Stefanova N, Jellinger KA, et al; Towards translational therapies for multiple system atrophy. Prog Neurobiol. 2014 Jul;118C:19-35. doi: 10.1016/j.pneurobio.2014.02.007. Epub 2014 Mar 2.
- Figueroa JJ, Singer W, Parsaik A, et al; Multiple system atrophy: prognostic indicators of survival. Mov Disord. 2014 Aug;29(9):1151-7. doi: 10.1002/mds.25927. Epub 2014 Jun 7.
Article History
The information on this page is written and peer reviewed by qualified clinicians.
Next review due: 15 Mar 2026
16 Mar 2021 | Latest version

Feeling unwell?
Assess your symptoms online for free