Behçet's disease
Peer reviewed by Dr Laurence KnottLast updated by Dr Colin Tidy, MRCGPLast updated 23 Mar 2022
Meets Patient’s editorial guidelines
- DownloadDownload
- Share
- Language
- Discussion
- Audio Version
- Add to preferred sources on Google
Medical Professionals
Professional Reference articles are designed for health professionals to use. They are written by UK doctors and based on research evidence, UK and European Guidelines. You may find the Behçet's disease article more useful, or one of our other health articles.
Synonym: Behçet's syndrome
What is Behçet's disease?
Behçet's disease is a complex multi-system disorder of unknown aetiology characteristically presenting with recurrent oral ulcers. It is presumed to be an autoimmune disease and includes involvement of the mucocutaneous, ocular, cardiovascular, renal, gastrointestinal, pulmonary, vascular, musculoskeletal, urological and central nervous systems. In many patients the activity of Behçet's disease diminishes with time.
Although the pathogenesis of Behçet's disease is unclear, some studies have shown that immunological aberrations play an important role in the development and progression of Behçet's disease. Infection-related trigger factors, including antigens and autoantigens, are believed to mediate the development of Behçet's disease in patients with a genetic predisposition, resulting in the production of numerous cytokines and chemokines to combat the infection-related factors.1
The International Classification Criteria of Behçet's disease are as follows:2
In the absence of other clinical explanations, patients must have recurrent oral ulceration (aphthous or herpetiform) observed by the physician or patient, recurring at least three times in a 12-month period, and two of the following:
Recurrent genital ulceration.
Eye lesions: anterior uveitis, posterior uveitis, cells in the vitreous by slit lamp examination or retinal vasculitis observed by an ophthalmologist.
Skin lesions: erythema nodosum, pseudo-folliculitis, papulopustular lesions or acneiform nodules in post-adolescent patients not on corticosteroids.
Pathergy (exaggerated skin injury occurring after minor trauma), read by a physician at 24-48 hours.
Behçet's disease epidemiology2 3
Behçet's disease is not commonly seen in Northern Europeans. The prevalence is highest in the Middle East, Mediterranean and Eastern Asia4 .
The prevalence of Behçet's disease is 80 to 370 cases per 100,000 population in Turkey, 10 per 100,000 in Japan and 0.6 per 100,000 in Yorkshire. European cases are more often described - but not exclusively - in the migrant population.
HLA-B51 is the most strongly associated known genetic factor. However, it accounts for less than 20% of the genetic risk, even in familial cases (less than 5%), which indicates that other genetic factors remain to be discovered. It is most common among patients aged 20-30 years.
An age of onset younger than 25 years is associated with a higher prevalence of eye disease and active clinical disease.
Males and females are equally affected. However, males tend to have a more active and more severe form of the disease.
Behçet's disease symptoms2 5
Nonspecific symptoms of Behçet's disease include tiredness, malaise, muscle pains, and transient fevers. Headaches can be common.
Although oral ulceration, genital ulceration and eye disease are the classic triad of manifestations, the cardiovascular, gastrointestinal, musculoskeletal and central nervous systems can also be affected6 .
Skin and mucous membranes:
Oral ulceration: the frequency of oral ulceration in Behçet's disease is thought to be 97-100%. Mouth ulcers can be painful and slow to heal, and therefore cause difficulties in eating, drinking, and speaking, leading to a reduction in quality of life.7
Oral ulceration in Behçet's disease
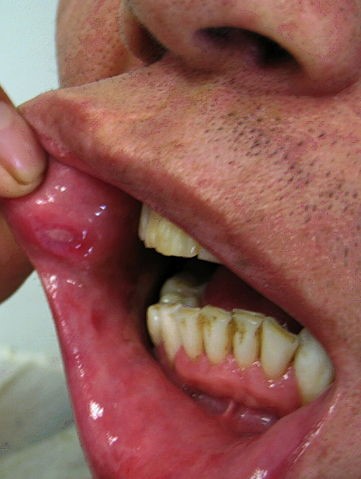
© Ahmet Altiner MD, Rajni Mandal MD, CC BY-SA 3.0, via Wikimedia Commons
Skin lesions can occur in the genital regions. The ulcers are very painful. They do not usually occur on the penis.
Painful nodules on the legs can occur due to erythema nodosum.
Patients often present with acne-like lesions on the arms and/or legs.
Eyes: about half of patients have uveitis. Other ocular manifestations include hypopyon, retinal vasculitis, retinal haemorrhage, blurred vision and photophobia.
Neurological: central nervous system (CNS) involvement occurs late in the disease. Memory impairment is the most common CNS manifestation. Impairment of balance, speech and movement can also occur. Thrombosis within the dural venous sinuses may occur. Peripheral nerve involvement is rare.8
Cardiovascular:
Vascular lesions include vasculitis of the small and large vessels, deep vein thrombosis and superficial thrombophlebitis, arterial occlusions and aneurysms.
Cardiac lesions include arrhythmias, pericarditis, vasculitis of the coronary arteries, endomyocardial fibrosis, and granulomas in the endocardium.
Arthritis: arthritis and arthralgias are common; a non-erosive arthritis occurs which most often affects the lower limbs, especially the knee. It can also affect the small joints. Back pain due to sacroiliitis may also occur.
Gastrointestinal: cramping abdominal pain, vomiting, diarrhoea or, less commonly, gastrointestinal bleeding can occur. Ulcerative lesions may occur in any part of the gastrointestinal tract.
Other manifestations include epididymitis, glomerulonephritis, lymphadenopathy, myositis and polychondritis. Amyloidosis may occur but is very uncommon.
Although the disease usually develops in young adulthood, it is reported that about 15-20% of all Behçet's patients develop the condition in childhood. Paediatric Behçet's disease differs from adult. Gastrointestinal system involvement, neurological findings, arthralgia and positive family history are more common in children, while genital lesions and vascular lesions are more common in adult patients. In addition, a better disease outcome with lower severity score and activity index has been reported in children.9
Differential diagnosis
This depends on which systems are involved. Many other possible conditions usually need to be considered in view of the very variable presentation.
Sarcoidosis, multiple sclerosis, Crohn's disease, Takayasu's arteritis, polychondritis or antiphospholipid syndrome need to be considered.2
Differential diagnosis of oral or genital ulcers includes herpes simplex, hand, foot and mouth disease, herpangina, histoplasmosis, inflammatory bowel disease, seronegative spondyloarthropathies, acne vulgaris and rheumatoid arthritis.10
Investigations
There is no relevant specific test for diagnosis. The ESR, CRP and other acute-phase reactants may be elevated during the acute phase and/or relapses but do not correlate with disease activity. Abnormalities of fibrinolysis, elevated factor VIII, immune circulating complexes and cryoglobulinaemia have been occasionally reported. Leukocytosis is frequently present. The positivity of HLA-B51 allele is of no diagnostic value. Cutaneous biopsy of intradermal injection with physiological saline solution may demonstrate vasculitis with immune complexes deposit.2
Investigations are therefore not required to confirm the diagnosis but are required when considering possible differential diagnoses and to assess the presenting features that may be present:
FBC may show mild anaemia and raised white cell count.
Nonspecific inflammatory markers: CRP, ESR, complement and acute-phase reactants may all be elevated during an acute attack.
Antiphospholipid antibodies are positive in 25% of patients.
Pathergy test: minor skin trauma induces an inflammatory papule or pustule after 24-48 hours. This is positive in up to 60% of patients.
Imaging studies, including CT and/or MRI scan, may be undertaken in some patients.
Angiography is required to identify and assess aneurysms.
Patients positive for HLA-B51 or HLA-B5 are more likely to be male and also have higher prevalences of genital ulcers, ocular manifestations and skin manifestations, and a decreased prevalence of gastrointestinal involvement.11
Behçet's disease treatment and management12 13
The aim of treatment is to prevent long-term damage. The most severe manifestation of Behçet's disease present in the patient usually determines the choice of treatment.
Local therapy:
Topical corticosteroids (eg, triamcinolone paste) are effective for oral or genital ulcerations if they are applied during the prodromal stage of ulceration when the symptoms are more mild.
Systemic manifestations:
Lesions resistant to local measures may require systemic treatment.
Systemic treatments are usually given for 1-2 years. They may be given longer for those with gastrointestinal involvement.
Patients with eye involvement need early and more aggressive treatment, usually with a brief course of corticosteroid with a longer-term immunosuppressant (eg, azathioprine).4
Patients with CNS involvement require the most aggressive treatment, usually with a corticosteroid plus either azathioprine, cyclophosphamide, or a TNF-alpha inhibitor.
Continual use of immunosuppressive medications may be required to suppress disease.
Corticosteroids are effective for acute manifestations but there is no evidence of any beneficial effect on disease progression.
Biotherapy approaches, including interferon-α (IFN-α), tumour necrosis factor-α (TNF-α) antagonists, and other agents that target interleukins and their receptors, have shown promising results in the treatment of patients with refractory Behçet's disease.1
Mucocutaneous lesions and joint involvement:
These may respond to non-steroidal anti-inflammatory drugs, prednisolone, levamisole, colchicine, dapsone, or sulfapyridine and thalidomide.
In most patients, arthritis can be managed with colchicine. Local corticosteroid injections may also be used.
Immunosuppressive therapy with azathioprine may be effective.
Major vessel disease:
Vasculitis is treated with immunosuppressive medication.
Prevention of acute deep vein thrombosis is by using immunosuppressive agents rather than anticoagulants.
If a venous thrombotic event does occur, however, warfarin is usually given.
For pulmonary and peripheral arterial aneurysms, immunosuppressants and corticosteroids are usually given.
Surgery
This may be required for:
Gastrointestinal complications - eg, perforation and peritonitis.
Aneurysms or ischaemic tissues affected by vasculitis or thrombosis.
Ventricular aneurysms and coronary thrombosis.
Eye involvement - eg, glaucoma, cataracts or retinal detachment.
CNS aneurysms or clots.
Complications
Vasculitis, rupture of aneurysms and thrombosis may all lead to potentially fatal cardiovascular complications.
CNS involvement can lead to permanent deficits.
Eye involvement may result in blindness.
Prognosis
There is a very variable course of recurrences and remissions lasting for years.
The majority of new manifestations occur within the first five years after onset of the disease.
Prognosis will depend on which systems are involved. Men tend to have a poorer prognosis.14 Mortality is usually low but death may occur as a result of neurological involvement, vascular disease, bowel perforation, cardiopulmonary disease, or as a complication of immunosuppressive therapy.
Although treatment of skin-mucosal manifestations, eye disease and pulmonary artery aneurysms has improved significantly in the past few decades, the treatment of CNS lesions is still problematic.15
Exclusive updates for healthcare professionals
Stay informed with the latest clinical updates, professional insights, and evidence-based guidance. The Patient Pro newsletter curates essential content for healthcare professionals—delivered straight to your inbox.
By subscribing you accept our Privacy Policy. You can unsubscribe at any time. We never sell your data.
Further reading and references
- Behcet's Syndrome Society
- Hatemi G, Seyahi E, Fresko I, et al; One year in review 2020: Behcet's syndrome. Clin Exp Rheumatol. 2020 Sep-Oct;38 Suppl 127(5):3-10. Epub 2020 Dec 10.
- Tong B, Liu X, Xiao J, et al; Immunopathogenesis of Behcet's Disease. Front Immunol. 2019 Mar 29;10:665. doi: 10.3389/fimmu.2019.00665. eCollection 2019.
- Saadoun D, Wechsler B; Behcet's disease. Orphanet J Rare Dis. 2012 Apr 12;7:20. doi: 10.1186/1750-1172-7-20.
- Leccese P, Alpsoy E; Behcet's Disease: An Overview of Etiopathogenesis. Front Immunol. 2019 May 10;10:1067. doi: 10.3389/fimmu.2019.01067. eCollection 2019.
- Fresko I, Yazici H; Treatment strategies for Behcet's disease. Expert Opin Pharmacother. 2008 Dec;9(18):3211-9.
- Nakamura K, Tsunemi Y, Kaneko F, et al; Mucocutaneous Manifestations of Behcet's Disease. Front Med (Lausanne). 2021 Feb 1;7:613432. doi: 10.3389/fmed.2020.613432. eCollection 2020.
- Ambrose NL, Haskard DO; Differential diagnosis and management of Behcet syndrome. Nat Rev Rheumatol. 2012 Sep 25. doi: 10.1038/nrrheum.2012.156.
- Taylor J, Glenny AM, Walsh T, et al; Interventions for the management of oral ulcers in Behcet's disease. Cochrane Database Syst Rev. 2014 Sep 25;(9):CD011018. doi: 10.1002/14651858.CD011018.pub2.
- Al-Araji A, Kidd DP; Neuro-Behcet's disease: epidemiology, clinical characteristics, and management. Lancet Neurol. 2009 Feb;8(2):192-204.
- Yildiz M, Haslak F, Adrovic A, et al; Pediatric Behcet's Disease. Front Med (Lausanne). 2021 Feb 3;8:627192. doi: 10.3389/fmed.2021.627192. eCollection 2021.
- Roett MA; Genital Ulcers: Differential Diagnosis and Management. Am Fam Physician. 2020 Mar 15;101(6):355-361.
- Maldini C, Lavalley MP, Cheminant M, et al; Relationships of HLA-B51 or B5 genotype with Behcet's disease clinical characteristics: systematic review and meta-analyses of observational studies. Rheumatology (Oxford). 2012 May;51(5):887-900. Epub 2012 Jan 11.
- EULAR recommendations for the management of Behçet disease; European League Against Rheumatism (January 2008)
- Alibaz-Oner F, Direskeneli H; Advances in the Treatment of Behcet's Disease. Curr Rheumatol Rep. 2021 May 20;23(6):47. doi: 10.1007/s11926-021-01011-z.
- Yazici H, Esen F; Mortality in Behcet's syndrome. Clin Exp Rheumatol. 2008 Sep-Oct;26(5 Suppl 51):S138-40.
- Yazici H, Fresko I, Yurdakul S; Behcet's syndrome: disease manifestations, management, and advances in treatment. Nat Clin Pract Rheumatol. 2007 Mar;3(3):148-55.
About the authorView full bio

Dr Colin Tidy, MRCGP
General Practitioner, Medical Author
MBBS, MRCGP, MRCP (Paediatrics), DCH
Dr Colin Tidy is an NHS Doctor, based in Oxfordshire.
About the reviewerView full bio

Dr Laurence Knott
General Practitioner, Medical Author
BSc (Hons) Biochemistry, MBBS
Dr Laurence Knott qualified in 1973 and has had extensive experience as a General Practitioner.
Article history
The information on this page is written and peer reviewed by qualified clinicians.
Article also available in English, German, Spanish, French, Italian, Portuguese, Hindi, Hebrew, Arabic, and Swedish.
Next review due: 22 Mar 2027
23 Mar 2022 | Latest version

Ask, share, connect.
Browse discussions, ask questions, and share experiences across hundreds of health topics.

Feeling unwell?
Assess your symptoms online for free
More in multisystem diseases
- Albinism
- Alpha-1-antitrypsin deficiency
- Antiphospholipid syndrome
- Chediak-Higashi syndrome
- Connective tissue diseases in pregnancy
- Discoid lupus erythematosus
- Edwards' syndrome
- Fatigue and TATT
- Granuloma annulare
- Granulomatosis with polyangiitis
- Heerfordt's syndrome
- Henoch-Schönlein purpura
- Hypermobility syndrome
- Kawasaki disease
- Mixed connective tissue disease
- Myositis - polymyositis and dermatomyositis
- Retinitis pigmentosa
- Systemic sclerosis
- Takayasu's arteritis
- Toxic shock syndrome