Retinitis pigmentosa
Peer reviewed by Dr Colin Tidy, MRCGPLast updated by Dr Rosalyn Adleman, MRCGPLast updated 27 Sept 2023
Meets Patient’s editorial guidelines
- DownloadDownload
- Share
- Language
- Discussion
- Audio Version
- Add to preferred sources on Google
Medical Professionals
Professional Reference articles are designed for health professionals to use. They are written by UK doctors and based on research evidence, UK and European Guidelines. You may find one of our health articles more useful.
What is retinitis pigmentosa?
The name retinitis pigmentosa (RP) was first applied by Dr Donders in 1857. It is the phenotypic description of several related, yet distinct, hereditary, progressive dystrophies of the photoreceptors of the retina and of the pigment epithelium (which lies just underneath the photoreceptors).
Patients present with ring scotoma and night vision problems, which progress to a slow loss of all peripheral vision; central vision is spared the longest. It is the leading cause of inherited retinal degeneration-associated sight impairment.
Pathology
RP is characterised by changes in pigment and arteriolar attenuation, often with some degree of optic nerve atrophy. Post-mortem examination has shown that the pigmentation is caused by cells from the pigment epithelium budding off and settling within the layers of the neural retina. In the late stages of RP a thinning of the retinal blood vessels is seen, probably resulting from the loss of many retinal cells reducing the need for blood.
The common end point is a gradual deterioration of the light-sensitive cells of the retina. Both rod and cone photoreceptors can be affected, the predominance of one over the other being determined by the particular genetic defect in that patient. Rod photoreceptor malfunction is the most commonly encountered problem in RP - cone dystrophies are distinct and present with a different set of problems.
There are various inheritance patterns. To date, more than 100 different genetic defects have been identified.1 These include the following: X-linked (5-15%), autosomal dominant (30-40%) and the remainder assumed autosomal recessive (50-60%).2 The autosomal dominant forms tend to have a milder course with a late, slow progression and preserved vision until the fifth or sixth decade. The X-linked form is the most severe; central vision is usually lost by the third decade. Isolated cases, with no family history, also commonly occur.1
How common is retinitis pigmentosa? (Epidemiology)
Prevalence in all ages is approximately 1 in 4,000, worldwide.3
There is geographical variation with a prevalence of 1 in 750 reported in India4 and 1 in 6500 in South Korea.5
Because of X-linked varieties, men may be affected slightly more than women.
Signs and symptoms of retinitis pigmentosa (presentation)
Symptoms
Three types of RP have been described, determined by age of onset.
The premature type occurs at a mean age of 7.5 years. There is a second type occurring around the age of 17 and a 'senile' type with onset in the fifties.
In one type of RP, Leber's amaurosis, babies may become severely sight impaired within the first six months of life.
Symptoms often start with impaired night vision (nyctalopia) or dark adaptation.
Progressive loss of peripheral vision is common (resulting in a tendency to trip over things), although there may be loss of central vision which tends to occur later. This eventually leads to impaired sight at a variable rate.
In some cases RP is first diagnosed following a road accident.
Signs
Typical features of RP can be seen on fundoscopy. Dispersion and aggregation of retinal pigment produce changes ranging from granules or mottling to distinctive focal aggregates with the appearance of bone spicules. The retina shows black or dark brown, star-shaped concentrations of pigmentation. The optic disc may appear pale and there may be vascular attenuation of the retinal vessels.
Fundus of patient with retinitis pigmentosa, mid stage
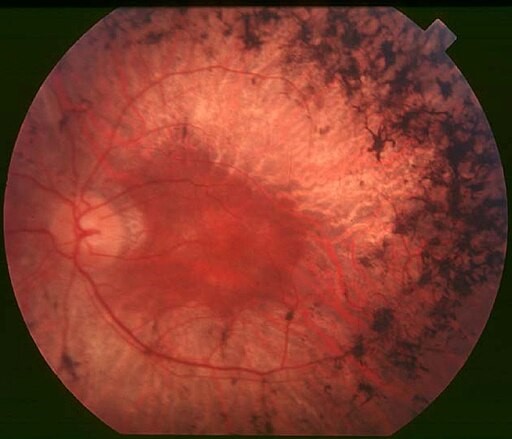
© Christian Hamel, CC BY 2.0, via Wikimedia Commons
There may be various patterns of change, including pigmentation limited to one quadrant of the retina, abnormalities which appear to be radiating out from the disc and changes associated with a severe vasculopathy. Associated ocular problems may include:
Myopia (frequently).
Subcapsular cataract.
Open-angle glaucoma (3% of patients).
Keratoconus.
Vitreous changes (most commonly a posterior vitreous detachment).
Systemic findings
RP is usually confined to the eye but may also be part of a syndrome with non-ocular features.6 At least 30 different associated syndromes have been identified.2
Patients with Usher syndrome have hearing loss, which may be profound or partial with a congenital or late onset. This accounts for about half of all cases of combined deafblindness.7 .
RP and hearing loss are also associated with:
Waardenburg's syndrome.8
Alström's syndrome.
Alport's syndrome.
Refsum's disease.
Other systemic conditions, all of which have their own systemic manifestations.
Short stature, renal dysfunction, and polydactyly are some signs of Bardet-Biedl syndrome or Laurence-Moon syndrome when associated with pigmentary retinopathy.
The mucopolysaccharidoses may be associated with RP (eg, Hurler's syndrome, Scheie's syndrome, Sanfilippo's syndrome), as well as the mitochondrial disorder, Kearns-Sayre syndrome, which manifests as ptosis, external ophthalmoplegia, and heart block.9
Differential diagnosis
Secondary pigmentary retinal degeneration occurs in a number of metabolic and neurodegenerative diseases, various syndromes and other eye diseases. In addition to those mentioned above, these include:
Friedreich's ataxia.
Mucopolysaccharidosis.
Muscular dystrophy (myotonic dystrophy).
Batten's syndrome.
Bassen-Kornzweig syndrome.
Homocystinuria.
Oxalosis.
Trauma.
Glaucoma with retinal pigment epithelial changes.
End-stage chloroquine retinopathy.
End-stage thioridazine retinopathy.
End-stage syphilitic neuroretinitis.
Cancer-related retinopathy.
Diagnosing retinitis pigmentosa (investigations)
Slit-lamp biomicroscopy is the key initial assessment. Further tests are to determine the functional integrity of the retina and optic nerve:
Visual acuity.
Visual field assessment.
Pupillary reflex response.
Colour defectiveness determination.
Refraction.
Intraocular pressure will also need to be measured. To find out more about these tests, see the separate Examination of the Eye article. Imaging includes:10
Retinal photography.
Ultrasound of the eye.
Fluorescein angiography.
Optical computer tomography (OCT).
All of these can be performed in a general clinic. The most critical diagnostic test is the electroretinogram (similar to EEG of the brain or ECG of the heart). It should be carried out in centres with the appropriate facilities (so, patients will need to be referred on if there is not one in their local hospital - this will be done by the ophthalmology team). It provides an objective measure of rod and cone function across the retina. It will typically show a marked reduction of rod and cone signals, although rod loss generally predominates.
Adaptive optics scanning laser ophthalmoscopy is a relatively new technique which can be used to detect early photoreceptor damage3 . Wide field fundus autofluorescence (FAF) is another newer imaging modality. It identifies areas of irregular distribution in the retina. Specific patterns of abnormal FAF are useful in the diagnosis of inherited retinal degenerations.11
Management of retinitis pigmentosa3
There is currently no approved therapy able to stop the evolution of RP or restore vision, so the current management aims to slow down the degenerative process, to provide low vision aids and to provide psychological support.12
However, significant advances have been made in recent years.
Various drugs have been proposed for the management but the evidence supporting their effectiveness is variable and generally limited.
Referral to a low vision specialist is very helpful.
Visual rehabilitation for patients with low visual acuity is helpful. It involves a multidisciplinary approach that focuses on the patient's functional abilities and needs.
Patients should make regular visits to an eye care specialist to screen for and treat any ocular complications such as cataracts, glaucoma and cystoid macular oedema.
The use of sunglasses to protect the retina from ultraviolet light may help preserve vision. Bright light can provoke the formation of free radicals which are damaging to the epithelium.
Genetic counselling is important and family members (siblings and offspring) should be examined for evidence of RP.
General counselling by experienced staff is vital. It is worth noting that most children will have enough sight to complete their education in normal schools.
The DVLA will need to be informed (by the patient) and there will be a requirement to do a specialised (Estermann) visual field test which is carried out by DVLA-approved optometrists; this is a legal requirement.
Eventually, registration for severe sight impairment or for sight impairment is required - see the separate Severe and Partial Sight Impairment article.
Drug
Vitamin A and fish oils: these were previously recommended but a Cochrane review demonstrated no clear benefits.13
Acetazolamide: in a small percentage of patients with RP, cystoid oedema may respond to oral carbonic anhydrase inhibitors, such as acetazolamide, with some subjective improvement in visual function. Topical carbonic anhydrase inhibitors like 1-2% dorzolamide or brinzolamide.
Lutein: this may slow retinal degeneration due to its antioxidant effect in animal models but benefits in humans have yet to be demonstrated.14
Bilberry: has been recommended by some practitioners of alternative medicine in doses of 80 mg. Animal studies have reported some benefit, although no controlled studies exist that document its safety or efficacy in treating human patients with RP.15
Immunosuppressive agents (including steroids): these have been used, with anecdotal success, in patients who present with anti-retinal antibodies.
Gene-specific and mutation-specific therapy: . Voretigene neparvovec, a recombinant adeno-associated virus vector carrying a normal copy of the RPE65 gene, has demonstrated improved vision in patients with Leber congenital amaurosis secondary to mutations in the RPE65 gene when injected into the subretinal space. It is approved in the UK for treatment of biallelic RPE65 mutation-associated retinal dystrophy.16 It is the first approved gene therapy for the eye in the UK. Clinical trials for gene therapy for Usher syndrome and X-linked RP are underway.1
Abetalipoproteinaemia (Bassen-Kornzweig syndrome): patients also have fat malabsorption. High levels of vitamin A may restore retinal function in early stages (vitamin E may also help).2
Refsum's disease: dietary reduction of phytanic acid can slow or halt retinitis in this condition.
Familial isolated vitamin E deficiency (alpha-tocopherol transport protein deficiency): treatment with vitamin E can halt disease progression.2
Surgical
Retinal pigment epithelium transplants are in the experimental phase.17 Other emerging therapies include retinal prostheses.
Where cataracts have occurred or significant keratoconus has developed, surgery for these conditions will also help.
Prognosis
The disorder will continue to progress, although slowly. Complete loss of vision is uncommon.
Prevention of retinitis pigmentosa
Some assessment of the risk of having an affected child may be made by genetic counselling. In the future, gene therapy and stem cell therapeutic strategies are likely to radically improve management of inherited retinal disease.18
Exclusive updates for healthcare professionals
Stay informed with the latest clinical updates, professional insights, and evidence-based guidance. The Patient Pro newsletter curates essential content for healthcare professionals—delivered straight to your inbox.
By subscribing you accept our Privacy Policy. You can unsubscribe at any time. We never sell your data.
Further reading and references
- Retinitis pigmentosa; Royal National Institute of Blind People (RNIB)
- Moreno ML, Merida S, Bosch-Morell F, et al; Autophagy Dysfunction and Oxidative Stress, Two Related Mechanisms Implicated in Retinitis Pigmentosa. Front Physiol. 2018 Jul 26;9:1008. doi: 10.3389/fphys.2018.01008. eCollection 2018.
- Nakamura N, Fujinami K, Mizuno Y, et al; Evaluation of cone function by a handheld non-mydriatic flicker electroretinogram device. Clin Ophthalmol. 2016 Jun 30;10:1175-85. doi: 10.2147/OPTH.S104721. eCollection 2016.
- Retinitis Pigmentosa; BMJ Best Practice, 2023
- Retinitis Pigmentosa; BMJ Best Practice, 2023
- Hartong DT, Berson EL, Dryja TP; Retinitis pigmentosa. Lancet. 2006 Nov 18;368(9549):1795-809.
- Verbakel SK, van Huet RAC, Boon CJF, et al; Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018 Sep;66:157-186. doi: 10.1016/j.preteyeres.2018.03.005. Epub 2018 Mar 27.
- Nangia V, Jonas JB, Khare A, et al; Prevalence of retinitis pigmentosa in India: the Central India Eye and Medical Study. Acta Ophthalmol. 2012 Dec;90(8):e649-50. doi: 10.1111/j.1755-3768.2012.02396.x. Epub 2012 May 18.
- Rim TH, Park HW, Kim DW, et al; Four-year nationwide incidence of retinitis pigmentosa in South Korea: a population-based retrospective study from 2011 to 2014. BMJ Open. 2017 May 9;7(5):e015531. doi: 10.1136/bmjopen-2016-015531.
- Ferrari S, Di Iorio E, Barbaro V, et al; Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics. 2011 Jun;12(4):238-49. doi: 10.2174/138920211795860107.
- Fahim A et al; Nonsyndromic Retinitis Pigmentosa Overview
- Kumar S, Rao K; Waardenburg syndrome: A rare genetic disorder, a report of two cases. Indian J Hum Genet. 2012 May;18(2):254-5. doi: 10.4103/0971-6866.100804.
- Fenzl CR, Teramoto K, Moshirfar M; Ocular manifestations and management recommendations of lysosomal storage disorders I: mucopolysaccharidoses. Clin Ophthalmol. 2015 Sep 7;9:1633-44. doi: 10.2147/OPTH.S78368. eCollection 2015.
- Mitamura Y, Mitamura-Aizawa S, Nagasawa T, et al; Diagnostic imaging in patients with retinitis pigmentosa. J Med Invest. 2012;59(1-2):1-11.
- Guidelines on Clinical Assessment of Patients with Inherited Retinal Degenerations; American Academy of Ophthalmology, 2022
- Sahni JN, Angi M, Irigoyen C, et al; Therapeutic challenges to retinitis pigmentosa: from neuroprotection to gene therapy. Curr Genomics. 2011 Jun;12(4):276-84. doi: 10.2174/138920211795860062.
- Schwartz SG, Wang X, Chavis P, et al; Vitamin A and fish oils for preventing the progression of retinitis pigmentosa. Cochrane Database Syst Rev. 2020 Jun 18;6(6):CD008428. doi: 10.1002/14651858.CD008428.pub3.
- Xue C, Rosen R, Jordan A, et al; Management of Ocular Diseases Using Lutein and Zeaxanthin: What Have We Learned from Experimental Animal Studies? J Ophthalmol. 2015;2015:523027. doi: 10.1155/2015/523027. Epub 2015 Nov 5.
- Osada H, Okamoto T, Kawashima H, et al; Neuroprotective effect of bilberry extract in a murine model of photo-stressed retina. PLoS One. 2017 Jun 1;12(6):e0178627. doi: 10.1371/journal.pone.0178627. eCollection 2017.
- Voretigene neparvovec for treating inherited retinal dystrophies caused by RPE65 gene mutations; NICE guidance, 2019
- Foik AT, Lean GA, Scholl LR, et al; Detailed Visual Cortical Responses Generated by Retinal Sheet Transplants in Rats with Severe Retinal Degeneration. J Neurosci. 2018 Dec 12;38(50):10709-10724. doi: 10.1523/JNEUROSCI.1279-18.2018. Epub 2018 Nov 5.
- Fahim A; Retinitis pigmentosa: recent advances and future directions in diagnosis and management. Curr Opin Pediatr. 2018 Dec;30(6):725-733. doi: 10.1097/MOP.0000000000000690.
About the authorView full bio

Dr Rosalyn Adleman, MRCGP
MRCGP
Dr Rosalyn Adleman, is an NHS GP working in north London.
About the reviewerView full bio

Dr Colin Tidy, MRCGP
General Practitioner, Medical Author
MBBS, MRCGP, MRCP (Paediatrics), DCH
Dr Colin Tidy is an NHS Doctor, based in Oxfordshire.
Article history
The information on this page is written and peer reviewed by qualified clinicians.
Article also available in English, German, Spanish, French, Italian, Portuguese, Hindi, Hebrew, Arabic, and Swedish.
Next review due: 25 Sept 2028
27 Sept 2023 | Latest version

Ask, share, connect.
Browse discussions, ask questions, and share experiences across hundreds of health topics.

Feeling unwell?
Assess your symptoms online for free
More in multisystem diseases
- Albinism
- Alpha-1-antitrypsin deficiency
- Antiphospholipid syndrome
- Behçet's disease
- Chediak-Higashi syndrome
- Connective tissue diseases in pregnancy
- Cystic fibrosis
- Eosinophilic granulomatosis with polyangiitis
- Graft-vs-host disease
- Hypermobility syndrome
- Kawasaki disease
- Libman-Sacks endocarditis
- Mixed connective tissue disease
- Relapsing polychondritis
- Seronegative arthropathies
- Sneddon's syndrome
- Syphilis
- Systemic lupus erythematosus
- Takayasu's arteritis
- Toxic shock syndrome