Retinoblastoma
Peer reviewed by Dr Hayley Willacy, FRCGP Last updated by Dr Mary Elisabeth Lowth, FRCGPLast updated 13 Sept 2016
Meets Patient’s editorial guidelines
- DownloadDownload
- Share
- Language
- Discussion
- Audio Version
- Add to preferred sources on Google
This page has been archived.
It has not been reviewed recently and is not up to date. External links and references may no longer work.
Retinoblastoma is an uncommon cancer of the eye which occurs in children under the age of 5. This leaflet describes retinoblastoma. It discusses the symptoms and signs of retinoblastoma and the management options children with retinoblastoma may be offered.
At a glance
Retinoblastoma is a rare eye cancer that mainly affects children under 5 years old.
It is caused by a genetic abnormality, which can be inherited or occur spontaneously.
A common first sign is a white pupil, noticeable in flash photographs.
Early diagnosis and treatment are crucial for successful outcomes.
Treatment options include freezing, heat, laser, plaque radiotherapy, chemotherapy, and surgery.
In developed countries, 99 out of 100 children diagnosed with retinoblastoma are cured.
Children with the inherited form need lifelong monitoring due to an increased risk of other cancers.
What is retinoblastoma?
Retinoblastoma is a tumour which usually affects children aged under 5. Most commonly, it affects children aged under 2, including newborn babies. It is a tumour of the nerve layer of the retina, which is the light-sensitive membrane at the back of the eye. The tumour grows from the retina very early in its life, when it is still developing.
See separate leaflet called Cancer - A General Overview for more general information about cancer
Side view of the structure of the eye
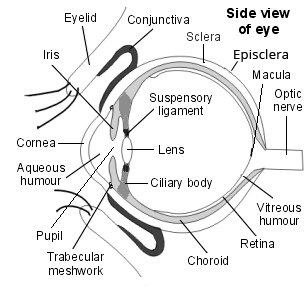
How common is retinoblastoma?
Retinoblastoma is very uncommon: it occurs in 1 in every 20,000 births. Around 80 cases are diagnosed in the UK each year. Even though retinoblastoma is a genetic disease, most of these cases occur in families who have never had a family member with retinoblastoma.
What causes retinoblastoma?
Retinoblastoma is a genetic disease. Genetic diseases are disorders of the 46 chromosomes which contain the coding (DNA) that our body needs in order to build and grow its cells. Retinoblastoma is caused by abnormalities on chromosome 13, which allow the tumour to develop. These abnormalities can be inherited from a parent, or happen for the first time at an early stage of development in the womb - this is called a mutation. If the mutation happens very early in the growth of the embryo it can be copied into all cells of the body. If it arises later in the emrbyo's development it may only occur in the eye.
9 out of 10 children who have the abnormal retinoblastoma gene will develop retinoblastoma..
Is retinoblastoma inherited?
In around 4 in every 10 retinoblastomas the abnormal gene is present in all the cells of the body, and can be passed on through inheritance. It is therefore called heritable retinoblastoma. The affected child may have an affected parent, or the abnormal gene may be a mutation that arose newly in the child whilst in the womb. However, as it is present throughout the body, the condition can be passed on to the affected child's children.
The remaining 6 out of every 10 retinoblastomas are caused by mutation in the genes of the cells in one eye only, so that the abnormal gene is not present elsewhere in the body. In this case the retinoblastoma cannot be passed on through inheritance and occurs in one eye only.
Everyone has two copies of chromosome 13. If you have heritable retinoblastoma one of those copies will be abnormal. When you conceive a child they will inherit only one copy of chromosome 13 from you and one from their other parent. Therefore, they have a 50/50 (1 in 2) chance of inheriting the abnormal gene.
Is retinoblastoma dangerous?
Yes - retinoblastoma is a cancerous (malignant) disease and, if untreated, it spreads within and outside the brain and is rapidly fatal. However, retinoblastoma treatment is highly effective if the disease is caught early. Early diagnosis and good access to treatment in the developed world means that 99 out of every 100 children diagnosed with retinoblastoma will be cured of the disease.
Unfortunately, in the developing world, where access to medical facilities and treatment options are often limited, retinoblastoma is usually discovered too late and more than 8 out of every 10 affected children will die from it.
What are the signs and symptoms of retinoblastoma?
Children who have family members with retinoblastoma may be diagnosed through screening before they have symptoms. If this is not the case, the first sign of retinoblastoma is often a white pupil that does not reflect light. This may be noticeable when a picture of your child is taken using flash photography - the pupil of the affected eye may look white in the photo.
Some children develop a squint. Some children are diagnosed as young babies because they do not appear to be developing normal vision. Less commonly, your child may have a painful, red or enlarged eye.
The heritable form of retinoblastoma tends to develop slightly earlier in life (at age 12 months or less) than the non-inherited type (more commonly diagnosed at age 24-30 months). It most often affects both eyes (although it can affect one only) and there may be other family members who are or have been affected. There can be several separate tumours in each eye and, very rarely, there are also tumours elsewhere in the brain.
What are the tests for retinoblastoma?
Diagnosis of retinoblastoma is usually made after an examination of your child's eye whilst they are under anaesthetic. The examination is performed by an eye specialist (ophthalmologist), who uses special magnifying tools to look into the eye through the pupil. Anaesthetic is needed, as very young children will not keep still enough to allow a detailed eye examination when they are awake. No sample (biopsy) is needed - retinoblastomas can be diagnosed by specialists just by their appearance.
When a retinoblastoma is diagnosed, your child may have tests to check the exact position and size of the tumour. This is known as staging and may include:
An ultrasound scan. This is a painless scan that uses sound waves to examine the eye and the surrounding area.
A magnetic resonance imaging (MRI) scan. This uses magnetism to build up a detailed picture of the eye and head.
A lumbar puncture. The doctor inserts a fine needle between the bones in the lower spine to remove a sample of the fluid from around the brain and spinal cord (cerebrospinal fluid). The fluid is examined to check whether there are any cancer cells present.
A blood test. This may be taken for genetic testing for the retinoblastoma gene (Rb1). The results of this test can take some months.
A bone scan, which is usually done if there is any suspicion that the tumour has spread into the bones, although this is unusual. Bone marrow samples and biopsies might be taken in this case.
What are the stages of retinoblastoma?
The retinoblastoma staging system divides the disease into:
Intraocular retinoblastoma (where there is disease in one or both eyes but no disease outside the eyes). This is divided into five grades, A to E, depending on the size and number of tumours, where exactly they are and where exactly they have spread to inside the eye.
Extraocular retinoblastoma (where the disease has spread outside the eye to surrounding tissues, or to elsewhere in the body).
What is the treatment for retinoblastoma?
The treatment of retinoblastoma depends on the stage of the disease at diagnosis. The first aim of treatment is to get rid of the cancer (and therefore save the life of your child). However, beyond this the secondary aims are to try to preserve vision in the eye.
Smaller tumours
Smaller tumours may be treated by local treatments to the eye itself. Several treatments may be needed. Treatments which may be used include:
Freezing treatment (cryotherapy).
Heat treatment (thermotherapy).
Laser treatment.
Plaque radiotherapy: a small radioactive disc is stitched over the tumour on the outside of the eye. It needs to stay in place for up to four days while the radiation destroys the cancer cells. Retinoblastoma cells are very sensitive to radiation.
Larger tumours
Larger tumours, or those that have spread beyond the eye, may require more complicated, combined treatments including:
Chemotherapy using anti-cancer drugs - those most commonly used to treat retinoblastoma are carboplatin, etoposide and vincristine. Chemotherapy is usually given into the circulation through a neck or arm vein. However, recently doctors have tried putting it directly into the blood vessel that supplies the affected eye.
Surgery: this was the traditional treatment and involved removal of the whole eye and its replacement with a cosmetic artificial eye (prosthesis). This treatment is called eye enucleation and is only offered if less drastic treatments are not expected to eradicate the tumour.
Radiotherapy: this is also called external beam radiotherapy. It can be given to the whole eye but it can affect surrounding tissue and is normally only used when other treatments have not worked well.
How are children with retinoblastoma followed up?
Following treatment, the eye specialist will examine your child's eye under anaesthetic and at frequent and regular intervals in order to check:
That the retina is healthy.
That the cancer has not come back.
That no new tumours have developed.
Follow-up is usually in a clinic for childhood cancers, called a paediatric oncology clinic. Appointments are likely to be several times a year, at least until your child is 6 or 7 years old.
Children with heritable retinoblastoma will also be given genetic counselling when they are old enough to understand it.
It is also important that parents - and later the children themselves - are aware of the increased risk of other cancers which they may face, and know what kind of symptoms to look out for (see below).
What are the long-term consequences of retinoblastoma?
The immediate side-effects of treatment, such as feeling sick (nausea) and tiredness, will usually stop after the treatment is complete. However, there are also possible longer-term effects after treatment of retinoblastoma:
Effects on the eye
Loss of vision. Most children will lose some vision after treatment of retinoblastoma, as they will be left with an area of abnormal retina. More than half of children have good or fairly good functional vision (with a visual acuity of at least 20/40). However, about 1 in 3 children will have very poor vision in the affected eye. If both eyes are affected this may mean the child has severe visual impairment.
If the eye has to be removed as a part of treatment then the eye socket (orbit) on that side may not grow as much as the other side, leading to a lack of symmetry. This can be largely prevented by use of correctly sized false eyes (eye implants).
Effects of chemotherapy
Carboplatin is commonly used in chemotherapy for retinoblastoma and this medicine can cause long-term hearing loss.
Risks of other cancers
Children who have had the heritable form of retinoblastoma have an increased risk of developing other cancers in their lifetime. Some of this increased risk is linked to radiation treatment, which is commonly used for retinoblastoma. However, an increased risk occurs even in patients who have not had radiation treatment. It appears likely that the gene abnormality that causes retinoblastoma also increases the likelihood of these other cancers developing. As as result of this, lifelong follow-up is needed and patients and their health carers need to be aware of what to look out for.
Other cancers
The most common second cancers to occur in heritable retinoblastoma survivors are sarcoma of bone or soft tissues, and skin cancer (melanoma). These are usually quite rare cancers but they are much more common in patients who have hereditary retinoblastoma. Secondary cancers can occur many years or even decades after the original retinoblastoma treatment. Around 1 in 100 hereditary retinoblastoma survivors will develop a secondary cancer every year. Health professionals and parents should always look carefully at complaints of bone pain or of developing lumps or changing moles in children who have had retinoblastoma.
Children with the non-heritable type of retinoblastoma - which is the most common type - are not thought to be at any greater risk of secondary cancers than anyone else. The abnormal genes in these children are only thought to be present in the affected eye and do not affect other parts of the body.
How do parents and carers cope with retinoblastoma?
For parents and carers, having your child diagnosed with cancer will be one of the worst situations you can imagine. You may feel overwhelmed with fear and anxiety, guilt, sadness, anger and uncertainty. You may feel that it must be your fault. You may worry that you aren't strong enough to support your child. These are all normal feelings and are part of the process that many parents go through at such a difficult time.
Your child may also have a variety of powerful fears and emotions, not only at the time treatment is needed but later too. The team treating your child will offer you help and support, and contacts of organisations that can help you through the process. Some of the organisations listed below can also be helpful.
The effects of looking after and supporting your family when your child has a life-threatening cancer can be overwhelming and can last for a very long time. Sometimes it is only after everything seems to be resolved that you really feel the stress and anxiety. If your child's treatment goes well, as it most often does in retinoblastoma, you may wonder why you are still haunted by your experience. However, this is a very normal human response. There are many organisations which can help and support you - some of the contact details are given below.
Can retinoblastoma be prevented?
Retinoblastoma is a genetic condition which cannot be prevented.
Prenatal testing is possible if hereditary retinoblastoma is present in your family - although the affected family member also needs to be tested in order to identify where exactly the family gene abnormality is found.
Prevention of secondary cancers is very important: limiting your child's lifelong exposure to agents which increase the likelihood of cancers (radiation, tobacco and ultraviolet light) may reduce the excess cancer risks. MRI scanning is preferred to computed tomography (CT) scanning where possible.
What is the outcome (prognosis) for retinoblastoma?
Retinoblastoma has the highest survival of all the childhood cancers in the UK. 99 out of every 100 children with retinoblastoma in the developed world survive this disease.
In developed countries, more than 9 out of every 10 of those who survive will have moderate-to-severe visual impairment, or will lose one or both eyes.
In developed countries it is very unusual for retinoblastoma to have spread throughout the body before diagnosis. In the developing world more than half of cases are very advanced when first diagnosed. Children whose retinoblastoma is very advanced have a much lower chance of survival. 87% of children with retinoblastoma worldwide die, mostly in developing countries. The survival chance is lowest in countries with the lowest incomes.
Should my family be screened for retinoblastoma?
If your child is thought to have the heritable form of retinoblastoma then other children in the family need to be screened regularly until they are between 3.5 and 5 years old.
Children of a parent who has had retinoblastoma, and siblings of an affected child, should undergo screening soon after birth. Regular screening with MRI is recommended. This is typically every six months for five years for those suspected of having heritable disease.
Genetic testing can identify children with heritable retinoblastoma if there is an affected family member to compare to.
Siblings of an affected child in a family in which no other family members have had the disease have only a small risk of disease.
See separate leaflet called Cancer - A General Overview for more general information about cancer
Dr Mary Lowth is an author or the original author of this leaflet.
Patient picks for Childhood cancers

Cancer
Neuroblastoma
Neuroblastoma is a cancer affecting developing nerve cells. Neuroblastoma is a rare cancer and most often occurs in children under 5 years old. Neuroblastoma often starts in the tummy (abdomen) but can spread to other parts of the body, especially bones, liver and skin. The treatments for neuroblastoma include surgery, chemotherapy and radiotherapy. The outcome (prognosis) depends on the stage of the neuroblastoma when it is first diagnosed.
by Dr Colin Tidy, MRCGP

Cancer
Rhabdomyosarcoma
A rhabdomyosarcoma is a type of soft tissue sarcoma. Rhabdomyosarcomas grow in the muscles of the body. Rhabdomyosarcomas can occur at any age but are much more common in children and only rarely affect adults. The treatments for rhabdomyosarcomas include surgery, chemotherapy or radiotherapy, or a combination of all three. Surgery may be used on its own for small localised tumours. About 2 in every 3 children with rhabdomyosarcoma will be cured with treatment. However, the outcome (prognosis) also depends on which part of the body is affected.
by Dr Colin Tidy, MRCGP
Frequently asked questions
What is the difference between heritable and non-heritable retinoblastoma?
Heritable retinoblastoma means the abnormal gene is present in all body cells and can be passed on to children. This form can affect both eyes and may have other affected family members. Non-heritable retinoblastoma is caused by a gene mutation only in cells of one eye, meaning it cannot be passed on and typically affects only one eye.
If my child has heritable retinoblastoma, what is the chance my future children will also have it?
If your child has heritable retinoblastoma, and you have the abnormal gene, your future children have a 50/50 (1 in 2) chance of inheriting the abnormal gene. This is because they will inherit one copy of chromosome 13 from you and one from the other parent.
How soon after birth should babies with a family history of retinoblastoma be screened?
Children of a parent who has had retinoblastoma, and siblings of an affected child, should undergo screening soon after birth. Regular screening with MRI is recommended, typically every six months for five years for those suspected of having heritable disease.
Why is anaesthesia used for eye examinations in children with suspected retinoblastoma?
Anaesthesia is needed because very young children will not keep still enough to allow a detailed eye examination when they are awake. This ensures the eye specialist can perform a thorough examination using special magnifying tools.
What is the likelihood of developing other cancers if my child had heritable retinoblastoma?
Children who have had the heritable form of retinoblastoma have an increased risk of developing other cancers in their lifetime. Around 1 in 100 hereditary retinoblastoma survivors will develop a secondary cancer every year. The most common secondary cancers are sarcoma of bone or soft tissues, and skin cancer (melanoma).
Further reading and references
- Improving outcomes with children and young people with cancer; NICE Cancer Service Guidance (2005)
- Retinblastoma; Contact a Family
- Retinoblastoma Treatment; National Cancer Institute
- Lohmann DR, Gallie BL; Retinoblastoma. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016. 2000 Jul 18 [updated 2015 Nov 19.
About the authorView full bio

Dr Mary Elisabeth Lowth, FRCGP
MA (Cantab), MB, BChir, DFFP, DRCOG, PG Cert, Med Ed, FRCGP, MA (London)
Dr Mary Lowth was a Suffolk GP for 20 years, specialising in paediatrics and child protection, and later in documentation of torture.
About the reviewerView full bio

Dr Hayley Willacy, FRCGP
General Practitioner, Medical Author
MBChB (1992), DRCOG, DFFP, MRCOG (Part 1) MRCGP (2007), DFSRH (2013), MSc - medical education (2020)
Dr Hayley Willacy was an NHS GP working in northwest England, who retired from clinical practice in 2022 after 30 years.
Article history
The information on this page is written and peer reviewed by qualified clinicians.
Article also available in English, German, Spanish, French, Italian, Portuguese, Hindi, Hebrew, Arabic, and Swedish.
13 Sept 2016 | Latest version

Ask, share, connect.
Browse discussions, ask questions, and share experiences across hundreds of health topics.

Feeling unwell?
Assess your symptoms online for free
Sign up to the Patient newsletter
Your weekly dose of clear, trustworthy health advice - written to help you feel informed, confident and in control.
By subscribing you accept our Privacy Policy. You can unsubscribe at any time. We never sell your data.
More in cancer
- Bone marrow biopsy and aspiration
- BRCA genes
- Breast screening
- Cancer of the uterus
- Cancer symptoms
- Cervical screening
- Children's cancers
- Gynaecological cancer
- Mammogram
- Mouth cancer
- Mucositis
- Neuroblastoma
- Non-melanoma skin cancer
- Ovarian cancer
- Paget's disease of bone
- Palliative care
- Prostate cancer
- Rhabdomyosarcoma
- Stages of cancer
- Testicular cancer