Malignant melanoma of skin
Peer reviewed by Dr Krishna Vakharia, MRCGPLast updated by Dr Colin Tidy, MRCGPLast updated 26 Aug 2022
Meets Patient’s editorial guidelines
- DownloadDownload
- Share
- Language
- Discussion
- Audio Version
- Add to preferred sources on Google
Medical Professionals
Professional Reference articles are designed for health professionals to use. They are written by UK doctors and based on research evidence, UK and European Guidelines. You may find the Melanoma skin cancer article more useful, or one of our other health articles.
See also the separate Malignant Melanoma article.
What is a malignant melanoma?
Normal melanocytes are found in the basal layer of the epidermis. Melanocytes are found in equal numbers in black and in white skin; however, the melanocytes in black skin produce much more melanin. People with dark brown or black skin are very much less likely to be damaged by ultraviolet (UV) radiation than those with white skin. Non-cancerous growth of melanocytes results in moles (benign melanocytic naevi) and freckles (ephelides and lentigines).
Most skin melanomas spread out within the epidermis. If all the melanoma cells are confined to the epidermis then the lesion is a melanoma in situ, which can be cured by excision because it has no potential to spread around the body. When the cancer has grown through the dermis it is known as invasive melanoma. Four clinical types of skin melanoma exist:
Lentigo maligna melanoma: a patch of lentigo maligna develops a papule or nodule, signalling invasive tumour.
Superficial melanoma: a large flat irregularly pigmented lesion which grows laterally before vertical invasion develops.
Malignant melanoma on thigh
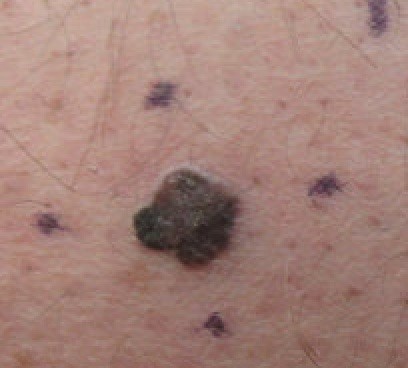
© Dermanonymous, CC BY-SA 4.0, via Wikimedia Commons
Nodular melanoma: the most aggressive type. It presents as a rapidly growing pigmented nodule which bleeds or ulcerates. Rarely, they are amelanotic (non-pigmented) and can mimic pyogenic granuloma.
Acral lentiginous malignant melanoma: arises as pigmented lesions on the palm, sole or under the nail and it usually presents late.
Once the melanoma cells have reached the dermis, they may spread to other tissues via the lymphatic system to the local lymph nodes or, via the bloodstream, to other organs. Metastases can occur virtually anywhere and at any time after a diagnosis of melanoma. Common sites for metastases are lymph nodes, liver, lung, bone and brain. In-transit metastases are deposits from a focus of cells moving along regional lymphatic channels.
How common is malignant melanoma1
Melanoma skin cancer is the 5th most common cancer in the UK, accounting for 4% of all new cancer cases (2016-2018). 50% of melanoma skin cancer cases in the UK are in females, and 50% are in males.
There are around 16,700 new melanoma skin cancer cases in the UK every year (2016-2018).
Incidence rates for melanoma skin cancer in the UK are highest in people aged 85 to 89 (2016-2018).
Each year more than a quarter (29%) of all new melanoma skin cancer cases in the UK are diagnosed in people aged 75 and over (2016-2018).
Since the early 1990s, melanoma skin cancer incidence rates have more than doubled (140%) in the UK. Rates in females have around doubled (106%), and rates in males have almost tripled (186%) (2016-2018).
In females, the most common specific location for melanoma skin cancers in the UK is the lower limb, while in males the most common specific location for melanoma skin cancers in the UK is the trunk (2016-2018).
Incidence rates for melanoma skin cancer are projected to rise by 7% in the UK between 2014 and 2035, to 32 cases per 100,000 people by 2035.
Melanoma skin cancer incidence rates in England in females are 52% lower in the most deprived quintile compared with the least, and in males are 54% lower in the most deprived quintile compared with the least (2013-2017).
Incidence rates for melanoma skin cancer are lower in the Asian and Black ethnic groups, compared with the White ethnic group, in England (2013-2017).
BRAF is a serine/threonine protein kinase activating the MAP kinase/ERK-signalling pathway. It is encoded on chromosome 7q34. Over 90% of melanomas contain the BRAFV600E mutation.2
Risk factors3
Factors that increase the risk of melanoma include:
Personal history or family history of skin cancer.
Pale skin (Fitzpatrick Skin Type I and II) that burns easily.
Red, blonde or light-coloured hair.
Blue or green eyes.
Unusually high sun exposure (the risk is higher with intermittent sun exposure than regular exposure).
History of sunburn, particularly blistering sunburn in childhood.
Use of tanning beds or sun beds, particularly if 10 or more sessions.
Large number of moles.
Increasing age from 15 years of age onwards.
Organ transplant recipients.
Malignant melanoma symptoms and differential diagnosis3
It can sometimes be difficult to differentiate melanoma from other lesions. Other types of pigmented lesions include:
Moles (naevi) - see the separate Intradermal and Compound Naevi, Spider Naevus, Junctional Naevus and Epidermal Naevus and its Syndromes articles.
Freckles.
Pigmented basal cell carcinoma.
Non-melanotic lesions which can be mistaken for melanoma include angiomas and haemangiomas, angiokeratomas, pyogenic granulomas, Kaposi's sarcoma and simple scabs.
See also the separate Black and Brown Skin Lesions article.
Referral4
Refer people (using a suspected cancer pathway referral for an appointment within two weeks) for melanoma if they have a suspicious pigmented skin lesion with a weighted 7-point checklist score of three or more:
Weighted 7-point checklist:
Major features of the lesions (scoring two points each):
Change in size.
Irregular shape.
Irregular colour.
Minor features of the lesions (scoring one point each):
Largest diameter 7 mm or more.
Inflammation.
Oozing.
Change in sensation.
Refer people (using a suspected cancer pathway referral for an appointment within two weeks) if dermoscopy suggests melanoma of the skin.
Consider a suspected cancer pathway referral (for an appointment within two weeks) for melanoma in people with a pigmented or non-pigmented skin lesion that suggests nodular melanoma.
Lesions which are suspicious for melanoma should not be removed in primary care.
Editor's note |
|---|
Dr Krishna Vakharia, 16th October 2023 Suspected cancer: recognition and referral4 The National Institute for Health and Care Excellence (NICE) has recommended that a person should receive a diagnosis or ruling out of cancer within 28 days of being referred urgently by their GP for suspected cancer. |
Malignant melanoma assessment5
Investigation is primarily by visual inspection and removal for histology where necessary. Diagnosis should be based on a full-thickness excisional biopsy.6
All pigmented skin lesions that are either referred for assessment or identified during follow-up in secondary or tertiary care should be assessed using dermoscopy. For clinically atypical melanocytic lesions that do not need excision at first presentation in secondary or tertiary care, baseline photography should be used with review after three months.
BRAF analysis of melanoma tissue samples from people with stage IIA or IIB primary melanoma should be considered, and should be performed on melanoma tissue samples from people with stage IIC to IV primary melanoma.
All pigmented lesions that are not viewed as suspicious of melanoma but are excised should have a lateral excision margin of 2 mm of clinically normal skin and cut to include subcutaneous fat. All excised lesions should be sent for histology.
Staging7
Staging of primary melanoma is based on the histological features of the lesion. Accurate staging is vital to determine appropriate treatment, follow-up and calculation of risk of recurrence. The American Joint Committee on Cancer (AJCC) staging system for cutaneous melanoma was revised in 2009.
Tumour:
TX: primary tumour cannot be assessed.
T0: no evidence of primary tumour.
Tis: tumour in situ.
T1: thickness 1 mm or less; a: without ulceration and mitosis <1/mm2; b: with ulceration or mitoses 1/mm2 or larger.
T2: thickness 1.01-2.00 mm; a: without ulceration; b: with ulceration.
T3: thickness 2.01-4.00 mm; a: without ulceration; b: with ulceration.
T4: thickness >4.00 mm; a: without ulceration; b: with ulceration.
Nodes:
NX: regional lymph nodes cannot be assessed.
N0: no nodal metastases.
N1: one metastatic node; a: micrometastasis (diagnosed after SLNB); b: macrometastasis (clinically detectable nodal metastases confirmed pathologically).
N2: 2-3 metastases; a: micrometastasis; b: macrometastasis; c: in-transit metastases/satellites without metastatic nodes.
N3: more than four metastatic nodes, or matted nodes, or in-transit metastases/satellites with metastatic nodes.
Metastases:
M0: no detectable evidence of distant metastases.
M1a: distant skin, subcutaneous, or nodal metastases.
M1b: lung metastases.
M1c: all other visceral metastases; any distant metastasis with elevated serum LDH.
The AJCC stages based on 'Tumour, Node, Metastasis' (TNM) classification are:
Stage 0 - Tis, N0, M0.
Stage IA - T1a, N0, M0.
Stage IB - T1b, N0, M0; T2a, N0, M0.
Stage IIA - T2b, N0, M0; T3a, N0, M0.
Stage IIB - T3b, N0, M0; T4a, N0, M0.
Stage IIC - T4b, N0, M0.
Stage III - any T, N 1-3, M0.
Stage IIIA - pathological (p) T1-4a, N1a, M0; pT1-4a, N2a, M0.
Stage IIIB - pT1-4b, N1a, M0; pT1-4b, N2a, M0; pT1-4a, N1b, M0; pT1-4a, N2b, M0; pT1-4a/b, N2c, M0.
Stage IIIC - pT1-4b, N1b, M0; pT1-4b, N2b, M0; any T, N3, M0.
Stage IV - any T, any N, M1a, M1b or M1c.
Staging investigations5
Imaging or sentinel lymph node biopsy (SLNB) should not be offered to people who have stage IA melanoma. Imaging should not be performed before SLNB unless lymph node or distant metastases are suspected.
SLNB should be considered for people who have melanoma with a Breslow thickness of 0.8 mm to 1.0 mm and at least one of the following features: ulceration, lymphovascular invasion, or a mitotic index of 2 or more. SLNB should be considered for people who have melanoma with a Breslow thickness greater than 1.0 mm.
Staging with whole-body and brain contrast-enhanced (CE)-CT should be considered for people with stage IIB melanoma, and should be offered to people with stage IIC to IV melanoma.
Staging with brain MRI, instead of brain CE-CT, should be considered if locally available and after discussion and agreement with the specialist skin cancer multidisciplinary team, and should be offered to children and young adults (from birth to 24 years) with stage IIB to IV melanoma, and women with stage IIB to IV melanoma who are pregnant.
Malignant melanoma treatment and management5
The primary treatment for malignant melanoma is wide local excision. Re-excision should be performed for proven melanomas if the margins are inadequate. Recommended surgical excision margins:
For some patients with advanced disease, management may consist entirely of supportive palliative care.
Vitamin D levels should be measured at diagnosis in all people with melanoma. People whose vitamin D levels are thought to be suboptimal should be given advice on vitamin D supplementation and monitoring.
Studies have recently been done looking at 'adjuvant' treatment. This is medication given after a main treatment to destroy any microscopic cancer cells and cut the chance of the cancer coming back.
In 2021, the National Institute for Health and Care Excellence (NICE) recommended that a drug called nivolumab should be recommended for some people with melanoma which had been completely cut out, as adjuvant treatment if there was evidence of spread to the lymph nodes.
In February 2022, NICE also recommended that another related drug, pembrolizumab, could be used in the same circumstances. This is because studies have shown that using pembrolizumab increases how long people live without the cancer coming back compared with people who do not have the treatment. It is not yet clear how much it increases how long people will live.
Managing stages 0-II melanoma
Excision:
Consider a clinical margin of at least 0.5 cm when excising stage 0 melanoma. Use a clinical margin of 1 cm when excising stage I melanoma (or when a 2 cm excision margin would cause unacceptable disfigurement or morbidity) and 2 cm when excising stage II melanoma.
Imiquimod for stage 0 melanoma:
Topical imiquimod should be considered to treat stage 0 melanoma in adults if surgery to remove the entire lesion with a 0.5 cm clinical margin would lead to unacceptable disfigurement or morbidity.
Consider a repeat skin biopsy for histopathological assessment after treatment with topical imiquimod for stage 0 melanoma, to check whether it has been effective.
Managing stage III melanoma
Completion lymphadenectomy:
Do not routinely offer completion lymph node dissection to people with stage III melanoma and micrometastatic nodal disease detected by SLNB unless there are factors that might make recurrent nodal disease difficult to manage, eg melanoma of the head and neck, people for whom stage III adjuvant therapies are contraindicated, or when regular follow-up is not possible.
Lymph node dissection:
Offer therapeutic lymph node dissection to people with palpable stage IIIB to IIID melanoma, or cytologically or histologically confirmed nodal disease detected by imaging.
Adjuvant systemic anticancer treatments:
Dabrafenib with trametinib is recommended as an option for the adjuvant treatment of resected stage III BRAF V600 mutation-positive melanoma in adults.8
Pembrolizumab is recommended as an option for the adjuvant treatment of completely resected stage 3 melanoma with lymph node involvement in adults.9
Nivolumab is recommended as an option for the adjuvant treatment of completely resected melanoma in adults with lymph node involvement or metastatic disease.10
Editor's note |
|---|
Dr Krishna Vakharia, 3rd November 2022 Pembrolizumab for adjuvant treatment of resected stage 2B or 2C melanoma11 NICE has recommended pembrolizumab as an option for the adjuvant treatment of completely resected stage 2B or 2C melanoma in people aged 12 years and over. Currently standard care is routine follow-up following a resection of stage 2B or 2C melanoma. Pembrolizumab as an adjuvant has been shown to increase how long people live without the cancer coming back. |
Adjuvant radiotherapy:
Do not offer adjuvant radiotherapy to people with stage IIIA melanoma. Do not offer adjuvant radiotherapy to people with resected stage IIIB to IIID melanoma unless a reduction in the risk of local recurrence is estimated to outweigh the risk of significant adverse effects.
Palliative treatment for in-transit metastases:
Offer surgery as the first option and if surgery is not feasible, or if the person has recurrent in-transit metastases, consider one of the following options based on their suitability for the person:
Systemic anticancer therapy.
Talimogene laherparepvec, in line with NICE's technology appraisal guidance on talimogene laherparepvec.
Isolated limb infusion or perfusion.
Radiotherapy
Electrochemotherapy.12A topical agent such as imiquimod.
Palliative treatment for superficial skin metastases:
Topical imiquimod should be considered to palliate superficial melanoma skin metastases.
Managing stage IV melanoma
Management of oligometastatic stage IV melanoma
Surgery or other ablative treatments (including stereotactic radiotherapy or radioembolisation) should be considered to prevent and control symptoms of oligometastatic stage IV melanoma in consultation with site-specific multidisciplinary teams (eg, for the brain or for bones).
Refer people with melanoma and brain metastases that might be suitable for surgery or stereotactic radiotherapy to the neuro-oncology multidisciplinary team for a recommendation about treatment.
Systemic anticancer treatment
Targeted treatments
Dabrafenib and vemurafenib are recommended as options for treating unresectable or metastatic BRAF V600 mutation-positive melanoma.13 14
Immunotherapy
Offer nivolumab plus ipilimumab to people with untreated stage IV or unresectable stage III melanoma if suitable.15
If nivolumab plus ipilimumab is unsuitable or unacceptable (for example, because of potential toxicity), offer pembrolizumab or nivolumab monotherapy.16 17
Targeted therapies for BRAF V600 mutation-positive melanoma
Encorafenib plus binimetinib, or dabrafenib plus trametinib should be offered to people with untreated BRAF-mutant stage IV or unresectable stage III melanoma if nivolumab plus ipilimumab, pembrolizumab, and nivolumab are contra-indicated or it is predicted there is not enough time for an adequate immune response (for example, because of high disease burden or rapid progression).18 19
If encorafenib plus binimetinib, and dabrafenib plus trametinib, are both unsuitable or unacceptable to the person, offer dabrafenib or vemurafenib to people for whom binimetinib and trametinib are contra-indicated.13 14
If targeted treatment is contra-indicated, consider treatment with chemotherapy (dacarbazine) or best supportive care.
For people with untreated BRAF-wild type stage IV or unresectable stage III melanoma for whom nivolumab plus ipilimumab, pembrolizumab, and nivolumab are contra-indicated, consider treatment with chemotherapy (dacarbazine) or best supportive care.
Editor's note |
|---|
Dr Krishna Vakharia, 15th February 2024 Nivolumab–relatlimab for untreated unresectable or metastatic melanoma in people 12 years and over20 NICE has recommended nivolumab–relatlimab as an option for untreated advanced (unresectable or metastatic) melanoma in people 12 years and over. It can only be used if it is stopped after 2 years of treatment, or earlier if the cancer progresses. Evidence shows that people on this treatment have longer before their cancer gets worse compared to those on nivolumab and pembrolizumab. It is thought to be as effective as treatment with nivolumab plus ipilimumab. Initial thoughts are that nivolumab–relatlimab can also keep people living longer compared to other treatments - but there is not much evidence to support this yet. |
Cytotoxic chemotherapy
For people with previously treated melanoma in whom immunotherapies and targeted therapies are contra-indicated, unsuitable or unacceptable, consider treatment with chemotherapy (dacarbazine) or best supportive care.
Do not routinely offer further cytotoxic chemotherapy to people with stage IV or unresectable stage III melanoma who have had previous treatment with dacarbazine except in the context of a clinical trial.
Palliative care
Refer people with incurable melanoma to specialist palliative care services for symptom management.
See also articles on palliative care.
Follow-up for patients with melanoma5
Offer follow-up for one year to people who have had stage IA melanoma, and for five years to people who have had stages IB to IV melanoma, using the table on follow-up after stages I to IV melanoma.
Routine follow-up after stages I to IV melanoma. Offer personalised follow-up to people with unresectable stage III or IV melanoma, people at increased risk of further primary melanomas, children and young adults, and women who are pregnant.
Stage IA:
Year 1: consider two clinic appointments, with discharge at the end of year 1. Do not routinely offer screening investigations (including imaging and blood tests) as part of follow-up.
Stage IB:
Year 1: offer two clinic appointments, and consider adding two ultrasound scans of the draining nodal basin if sentinel lymph node biopsy (SLNB) was considered but not done.
Years 2 and 3: offer one clinic appointment each year, and consider adding one ultrasound scan of the draining nodal basin each year if SLNB was considered but not done.
Years 4 and 5: offer one clinic appointment each year. Discharge at the end of year 5.
Stage IIA:
Years 1 and 2: offer two clinic appointments each year, and consider adding two ultrasound scans of the draining nodal basin each year if SLNB was considered but not done.
Year 3: offer one clinic appointment, and consider adding one ultrasound scan of the draining nodal basin if SLNB was considered but not done.
Years 4 and 5: offer one clinic appointment each year. Discharge at the end of year 5.
Stage IIB:
Years 1 and 2: offer four clinic appointments each year, and consider two whole-body and brain contrast-enhanced CT (CE-CT) scans each year. Consider adding two ultrasound scans of the draining nodal basin each year if SLNB was considered but not done.
Year 3: offer two clinic appointments and consider two whole-body and brain CE-CT scans. Consider adding two ultrasound scans of the draining nodal basin if SLNB was considered but not done.
Years 4 and 5: offer one clinic appointment each year and consider one whole-body and brain CE-CT scan each year. Discharge at the end of year 5.
Stage IIC:
Years 1 and 2: offer four clinic appointments and two whole-body and brain CE CT scans each year. Consider adding two ultrasound scans of the draining nodal basin each year if SLNB was considered but not done.
Year 3: offer two clinic appointments and two whole-body and brain CE-CT scans. Consider adding two ultrasound scans of the draining nodal basin if SLNB was considered but not done.
Years 4 and 5: offer one clinic appointment and one whole-body and brain CE-CT scan each year. Discharge at the end of year 5.
Stage IIIA to IIIC not currently having adjuvant therapy:
Years 1 to 3: offer four clinic appointments and two whole-body and brain CE-CT scans each year. Consider adding two ultrasound scans of the draining nodal basin each year if the person has a positive sentinel lymph node.
Years 4 and 5: offer two clinic appointments and one whole-body and brain CE-CT scan each year. Discharge at the end of year 5.
IIID and resected IV not currently having adjuvant therapy:
Years 1 to 3: offer four clinic appointments and four whole-body and brain CE-CT scans each year.
Years 4 and 5: offer two clinic appointments and two whole-body and brain CE-CT scans each year. Discharge at the end of year 5.
IIIA to IIIC, IIID and resected IV having adjuvant therapy:
During adjuvant therapy, base follow-up on therapeutic requirements.
Prognosis1 3 6
Melanoma, especially when diagnosed at an advanced stage, can cause serious morbidity and may be fatal despite treatment. Melanoma has the greatest potential for metastasis compared with other skin malignancies. Therefore it has the highest incidence of mortality.
Prognosis is dependent on the stage of the melanoma, which depends on the thickness, level of ulceration and spread to local lymph nodes. The five-year survival is about 95% for people with stage 1A melanoma (less than 1 mm thickness; no ulceration; mitoses less than 1 mm-2), compared with 7-19% of people with stage 4 melanoma (spread to distant lymph nodes or other parts of the body).
Almost all (98.2%) of people diagnosed with melanoma skin cancer in England survive their disease for one year or more.
Around 9 in 10 (91.3%) of people diagnosed with melanoma skin cancer in England survive their disease for five years or more.
It is predicted that almost 9 in 10 (87.4%) of people diagnosed with melanoma skin cancer in England survive their disease for ten years or more.
Melanoma survival rates are higher in women.
Survival among melanoma patients decreases with increasing age. The melanoma mortality rate has increased over a period of 25 years for men but not for women.6
Melanoma patients also have increased risks for other skin tumours. In patients with lentigo maligna melanomas. In one study, 35% of patients developed another cutaneous malignancy within five years.6
Malignant melanoma prevention
A reduction in people's exposure to sun has not led to a significant reduction in the incidence of melanoma and sun avoidance may be detrimental (eg, as a cause of vitamin D deficiency).
Although large-scale primary prevention programmes, such as public health education campaigns aimed at reducing exposure to sun, may lead to reduced incidence, such programmes have not yet been proved effective.
Avoiding sunburn and excessive sun exposure without protection seems to be the most important message in skin cancer prevention without advocating keeping away from the sun altogether.
Sunbed use: current recommendations are that sunbeds should be avoided, especially in relation to premature skin ageing. Individuals with red hair and freckles, or multiple atypical naevi, should avoid sunbeds since their risks of developing both melanoma and non-melanoma skin cancer are already significantly increased.
Secondary prevention with early detection of melanoma saves lives. Educating the public and health professionals to recognise melanoma early is crucial. Rapid referral for surgery and further management are imperative to improve outcomes.
Individuals with multiple atypical naevi, a family history of melanoma and/or multiple cancers should be referred to a dermatologist for screening, as their risk of melanoma is significantly increased.
Exclusive updates for healthcare professionals
Stay informed with the latest clinical updates, professional insights, and evidence-based guidance. The Patient Pro newsletter curates essential content for healthcare professionals—delivered straight to your inbox.
By subscribing you accept our Privacy Policy. You can unsubscribe at any time. We never sell your data.
Further reading and references
- Sunlight exposure: risks and benefits; NICE Guidance (February 2016)
- Cutaneous melanoma; Scottish Intercollegiate Guidelines Network (January 2017)
- Cutaneous Melanoma: Clinical Practice Guidelines; European Society for Medical Oncology (2019)
- Cabrera R, Recule F; Unusual Clinical Presentations of Malignant Melanoma: A Review of Clinical and Histologic Features with Special Emphasis on Dermatoscopic Findings. Am J Clin Dermatol. 2018 Nov;19(Suppl 1):15-23. doi: 10.1007/s40257-018-0373-6.
- Testori AAE, Ribero S, Indini A, et al; Adjuvant Treatment of Melanoma: Recent Developments and Future Perspectives. Am J Clin Dermatol. 2019 Dec;20(6):817-827. doi: 10.1007/s40257-019-00456-4.
- Skin cancer incidence statistics; Cancer Research UK
- Kozyra P, Krasowska D, Pitucha M; New Potential Agents for Malignant Melanoma Treatment-Most Recent Studies 2020-2022. Int J Mol Sci. 2022 May 29;23(11). pii: ijms23116084. doi: 10.3390/ijms23116084.
- Melanoma and pigmented lesions; NICE CKS, July 2022 (UK access only)
- Suspected cancer: recognition and referral; NICE guideline (2015 - last updated April 2026)
- Melanoma: assessment and management; NICE Guidance (July 2015 - last updated July 2022)
- Cutaneous Melanoma: Clinical Practice Guidelines; European Society for Medical Oncology (2019)
- Balch CM, Gershenwald JE, Soong SJ, et al; Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009 Dec 20;27(36):6199-206. Epub 2009 Nov 16.
- Dabrafenib with trametinib for adjuvant treatment of resected BRAF V600 mutation-positive melanoma; NICE Technology appraisal guidance, October 2018
- Pembrolizumab for adjuvant treatment of completely resected stage 3 melanoma; NICE Technology appraisal guidance, February 2022
- Nivolumab for adjuvant treatment of completely resected melanoma with lymph node involvement or metastatic disease; NICE Technology appraisal guidance, March 2021
- Pembrolizumab for adjuvant treatment of resected stage 2B or 2C melanoma; NICE Technology appraisal guidance, October 2022
- Electrochemotherapy for metastases in the skin from tumours of non-skin origin; NICE Interventional procedure guidance, March 2013
- Dabrafenib for treating unresectable or metastatic BRAF V600 mutation‑positive melanoma; NICE Technology Appraisal Guidance, October 2014
- Vemurafenib for treating locally advanced or metastatic BRAF V600 mutation‑positive malignant melanoma; NICE Technology appraisal guidance, December 2012 - last updated January 2015
- Nivolumab in combination with ipilimumab for treating advanced melanoma; NICE Technology appraisal guidance, July 2016
- Pembrolizumab for advanced melanoma not previously treated with ipilimumab; NICE Technology appraisal guidance, November 2015
- Nivolumab for treating advanced (unresectable or metastatic) melanoma; NICE Technology appraisal guidance, February 2016
- Encorafenib with binimetinib for unresectable or metastatic BRAF V600 mutation-positive melanoma; NICE Technology appraisal guidance, February 2019
- Trametinib in combination with dabrafenib for treating unresectable or metastatic melanoma; NICE Technology appraisal guidance, June 2016
- Nivolumab–relatlimab for untreated unresectable or metastatic melanoma in people 12 years and over; NICE Technology appraisal guidance, February 2024
About the authorView full bio

Dr Colin Tidy, MRCGP
General Practitioner, Medical Author
MBBS, MRCGP, MRCP (Paediatrics), DCH
Dr Colin Tidy is an NHS Doctor, based in Oxfordshire.
About the reviewerView full bio

Dr Krishna Vakharia, MRCGP
Chief Medical Officer for Health, Optum UK
MBChB, MRCGP(2013), BMedSci (hons), DFSRH, DRCOG, PGDipDerm (Distn)
Dr Krishna Vakharia is an NHS GP. She is also a regular examiner for the postgraduate Diploma in Practical Dermatology at Cardiff University as well as being the Chief Medical Officer for health at Optum UK.
Article history
The information on this page is written and peer reviewed by qualified clinicians.
Article also available in English, German, Spanish, French, Italian, Portuguese, Hindi, Hebrew, Arabic, and Swedish.
Next review due: 25 Aug 2027
26 Aug 2022 | Latest version

Ask, share, connect.
Browse discussions, ask questions, and share experiences across hundreds of health topics.

Feeling unwell?
Assess your symptoms online for free
More in dermatology
- Antifungal medications
- Basal cell carcinoma
- Blind treatment of bacterial infection
- Buruli ulcer
- Cellulitis and erysipelas
- Coxsackievirus infection
- Eczema on hands and feet
- Febrile neutrophilic dermatosis
- Genital herpes in pregnancy
- Granuloma annulare
- Haemangiomata of skin
- Head lice
- Lipodystrophy syndrome
- Mastocytosis and mast cell disorders
- Mycobacterial skin infections
- Myositis - polymyositis and dermatomyositis
- Parvovirus infection
- Pemphigus
- Pompholyx