Ehlers-Danlos syndrome
Peer reviewed by Dr Colin Tidy, MRCGPLast updated by Dr Rachel Hudson, MRCGPLast updated 8 Oct 2024
Meets Patient’s editorial guidelines
- DownloadDownload
- Share
- Language
- Discussion
- Audio Version
- Add to preferred sources on Google
The Ehlers-Danlos syndromes (EDS) are a group of conditions that affect the stretchiness and strength of supporting tissues in the body, including skin, joints, blood vessels and internal organs. They vary in their impact from very mild through to very severe.
At a glance
Ehlers-Danlos syndromes (EDS) are a group of conditions affecting connective tissue, usually inherited.
There are 13 types of EDS, with symptoms varying from mild to severe, even life-threatening.
Common symptoms include unusually stretchy and fragile skin, and very flexible joints.
Some types can also affect blood vessels, digestion, eyes, teeth, and heart valves.
Diagnosis is often based on symptoms, as genetic tests are not available for all types.
Treatment aims to manage symptoms and protect skin and joints, as there is no cure.
Outlook varies; the rare vascular type can cause life-threatening complications.
What are the Ehlers-Danlos syndromes?
The Ehlers Danlos syndromes (EDS) are a group of conditions which are usually (although not always) inherited from parents. EDS affects connective tissue - people with EDS have a problem with the formation and structure of connective tissue in the body. Connective tissue is a particularly important component of skin, muscles and ligaments, blood vessels and heart valves.
Whilst the symptoms of EDS may be mild and undiagnosed, for some people with EDS the symptoms can be severe, life-changing and even life-threatening.
There are 13 types of EDS. Most forms of EDS affect:
The skin (which can be unusually stretchy and sometimes fragile).
The joints which can be 'double-jointed' (hypermobile).
Some types also cause general symptoms such as:
Tiredness.
Pain.
Poor sleep.
Some affect other parts of the body, including:
Blood vessels.
The digestive system.
The eyes.
The teeth.
The heart valves.
The type of EDS inherited from parents is always the same type - for example, a parent with vascular EDS cannot pass on hypermobile EDS to their child. These can be inherited via autosomal dominant inheritance (for hypermobile, classical and venous EDS) which means one in two children will be affected on average, or autosomal recessive inheritance (for kyphoscoliotic EDS) which means one in four children will be affected on average. Occasionally EDS is not inherited from a parent but has been caused by a random gene mutation.
Ehlers-Danlos syndrome or syndromes?
Until fairly recently, the name 'Ehlers-Danlos syndrome' was used to describe all forms of the condition. However, there are many different types of EDS and EDS is now therefore called 'the Ehlers-Danlos syndromes'.
How common are the Ehlers-Danlos syndromes?
It is estimated that EDS can occur in between 1 in 5,000 and 1 in 250,000 births.
The Ehlers-Danlos syndromes symptoms
Symptoms range from mild to very severe, and vary with type of EDS.
Milder forms of EDS are often not diagnosed until early adulthood, as it can take some time before the symptoms and signs become noticeable.
The more severe types, however, are diagnosed mainly in childhood. Some types are so rare that it is hard to generalise about normal symptoms. Below are the main symptoms seen in EDS.
Hypermobility syndrome is a condition where there are hypermobile joints but none of the other EDS symptoms. They are often treated in the same way.
Joint changes
Joints are loose and 'double-jointed' (hypermobile), so that they have a greater than normal range and degree of movement. This occurs in all types of EDS.
Ehlers-Danlos syndrome finger hyperreflexia

© Piotr Dołżonek, CC BY-SA 4.0, via Wikimedia Commons
Dislocations of joints may easily occur, and people with EDS can often put their own joints back into place because the tissues are so stretchy. Joint pains are also common.
Skin changes
In all types of EDS except the vascular type, the skin feels soft and is unusually stretchy.
Ehlers-Danlos syndrome hyperelastic skin
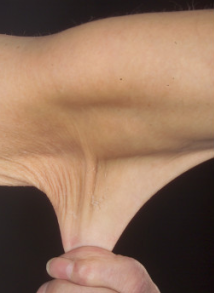
© Whitaker JK, Alexander P, Chau DY, Tint NL, CC BY 2.5, via Wikimedia Commons
The picture shows how easy it is to pull the skin downwards and away from the body. When let go, the skin immediately returns to its normal position.
Many affected people find that their skin splits and bruises easily and that skin injuries heal slowly. People with some types of EDS have scars that tend to become wide and thin. Blood vessels near to the skin surface can be very delicate and can be damaged easily.
Mobile lumps can develop under the skin of people with EDS, particularly around the elbows and knees, and tiny hard lumps may also form around the heel.
In the vascular type of EDS, the skin is not stretchy but it may look slightly transparent, so that blood vessels can be seen underneath the skin.
Muscles and skeleton
The muscles of people with many forms of EDS tend to be floppier and less strong than those of people without EDS. In severe cases, found in childhood, this may be noticed because of difficulty with walking, such as falling over frequently, or because of problems with balance. In more severe cases, people find it helpful to use a stick or a wheelchair to help them get about.
Poor grip and difficulty writing are sometimes a problem. Special aids may be needed at school, in the kitchen and in other places where grip is important. Sometimes the voice tires easily when shouting, singing or talking loudly. Easy tiring of the muscles is common.
People with EDS are more prone to hernias in the groin and in surgical scars. In some types of EDS, curvature of the spine (scoliosis) can develop.
Tiredness and sleep disturbance
Tiredness, getting tired easily after physical activity (fatiguability) and dizziness are very common in EDS of many types. Sleep disorders are also common. The milder types of EDS can seem similar to, and is often mistaken for, chronic fatigue syndrome and/or fibromyalgia.
Pain
Widespread pain or pain in the limbs is a common feature of most types of EDS. Exercise is important but should be chosen to minimise the risks of injuries - swimming, walking, tai chi or using a stationary bike are all good options.
Mood
Difficulties with mood, particularly anxiety, panic disorder and depression, are common conditions in the general population but they are more commonly experienced by people with EDS. This is thought mainly to be due to the constant high risks of joint dislocation (with the associated pain and disability) with little or no warning.
Heart
An uncomfortable feeling of thudding of the heart (palpitations) and a faster heart rate are seen in hypermobile EDS. It is also more common to experience dizziness and a faster pulse rate on standing in hypermobile EDS.
Some people with EDS have problems with their heart valves due to connective tissue changes, particularly as they get older, and they may develop a murmur.
In the rare vascular type of EDS, serious complications involving blood vessels can develop, including aneurysms and blood vessel rupture. This risk can be lowered by medication.
Lungs
Asthma-like symptoms can occur in some forms of EDS The exact mechanism for this is unclear.
Digestive system
Constipation and diverticulosis (in which there are bulges in the wall of the large intestine) are more common in people with EDS.
Hiatus hernia and irritable bowel syndrome are also more common than in the general population.
Many people with EDS experience 'heartburn' (dyspepsia).
Nausea is commonly noticed.
People with EDS are more than usually prone to travel sickness.
Prolapse of the bowel (rectal prolapse can occur) is quite common.
Bowel rupture is a possible (rare) complication of vascular EDS.
Eyes, ears and teeth
Tinnitus is common in several types of EDS. Many people also have dizziness (vertigo).
Some very rare types of EDS affect the eyes. The areas around the pupils (sclera) may be blue rather than white and the eyeballs themselves may be very fragile. One rare subtype also affects the gums and teeth.
Children's health
Children with EDS often show delay in crawling, walking and standing. They tend to be more floppy than usual. Some rare types of EDS can affect growth and cause early joint dislocations.
Women's health
Prolapse of the womb (uterus) is more common in EDS. Prolapse is a common condition in women who have children; however, it is seen in EDS in women who have not had children more commonly than you would otherwise expect.
Causes of the Ehlers-Danlos syndromes
The syndromes are genetic conditions. The genes which cause most types of EDS have been clearly identified and can be tested for. However, the most common form of EDS, hypermobility EDS (hEDS), is an exception to this, as the gene which causes it has not yet been identified.
In some cases, the faulty gene is inherited from an affected parent. However, a person may also be affected if there is a new mutation in the gene, in which case EDS may occur in people with no medical history of the disorder in their family.
The more common types of EDS show autosomal dominant inheritance, meaning having one faulty gene, from one parent, will cause the condition (and the parent will also have the condition). Rarer types of EDS show autosomal recessive inheritance.
How do the Ehlers Danlos syndromes affect pregnancy?
Problems that can occur when pregnant
These include:
Varicose veins, haemorrhoids and vulval varicosities.
Reflux, which tends to get worse: many people with EDS have ongoing reflux and symptoms may worsen during pregnancy.
Swelling of legs and hands, which is common in pregnancy but tends to be worse in EDS.
Carpal tunnel syndrome, which is common in pregnancy in EDS.
Women with EDS - may be more than usually prone to morning sickness.
Many people with EDS experience regular headaches and these tend to get worse in early pregnancy.
Many people with EDS have tinnitus due to instability of the bones in the middle ear. Pregnancy may exacerbate this condition.
Women with EDS who need to protect their joints throughout pregnancy.
Women with EDS who often have worse aches and pains in pregnancy, including:
Back pain.
Generalised aches and pains.
Palpitations and extra heartbeats, which may increase in pregnancy.
Breast changes which occur in most pregnant women. The stretchy skin of EDS means that extra support is important. People with EDS are more prone to stretchmarks generally.
Anxiety and depression can be made worse by pregnancy.
Problems that can occur during delivery
These include:
Early breaking of the waters - preterm rupture of membranes (PROM).
Women with EDS sometimes have a poor response to lidocaine (used in epidurals). Review by an anaesthetist before labour is advised to discuss likely effective options.
Delivery of the baby can be fast in EDS because the tissues 'give' easily and don't resist pushing the baby out. It's important for to try midwives to keep delivery slow and controlled.
Abnormal position of the baby is more common as the baby is not held so tightly by the tissues but this does not generally make labour more difficult in EDS.
Rarely, the womb (uterus) can rupture during labour.
Problems that can occur after delivery
These include:
Bleeding after delivery.
Fragile skin is more prone to tearing and may take longer to heal.
Dissolvable stitches may dissolve too early so silk sutures may be recommended.
How are the Ehlers-Danlos syndromes diagnosed?
EDS is usually suspected because of the typical symptoms of one of the types. The doctor may ask about close family members and any problems they may have had with their skin or joints. Once the condition is suspected, specialist testing, including genetic counselling and testing, may be offered to determine the type of EDS.
The exception to this is double-jointed (hypermobility) EDS (hEDS), the most common type, for which there is so far no genetic test. The diagnosis of hEDS is made from discovering a very specific set of symptoms. This is described under the description of hEDS below.
Treatment for the Ehlers-Danlos syndromes
There is no cure for EDS. Treatment is aimed at trying to protect the skin and the joints from further damage and at managing other symptoms which arise. For most people with EDS, generally speaking:
Physiotherapy can be very helpful to build strength and help balance, as well as to provide support for joints by strengthening muscles.
The 'talking therapy' known as cognitive behavioural therapy (CBT) is often recommended for anyone living with a chronic condition that affects their life.
For patients with fragile skin, injuries should ideally be stitched by a plastic surgeon who understands EDS, and stitches should be left in twice as long as usual.
Anti-inflammatory tablets and analgesics such as paracetamol may be required to control pain.
More detail about managing EDS is given under the description of each type, below.
What is the outlook for the Ehlers-Danlos syndromes?
The outlook (prognosis) is variable and depends on the type of EDS. Most forms of EDS do not affect a person's lifespan and people with milder symptoms are able to live their lives without any restrictions. The need to protect the skin and joints in those with more marked symptoms can limit a person's chosen activities - particularly having to reduce activities with a risk of physical injury, such as rugby - and can cause significant disability.
Many people with EDS have more severe symptoms and may have significant disabilities and therefore a severe restriction of their lives.
If someone has the rare type of EDS that affects the blood vessels (the vascular type) then there is a risk of bursting (rupture) of large blood vessels or internal organs, and therefore premature death.
Types of Ehlers-Danlos syndromes
The classification of EDS into different types has changed several times over the years. In 2017 there was a major change to the current classification. This divides EDS into 13 types.
These are:
Hypermobile Ehlers-Danlos syndrome.
Classical Ehlers-Danlos syndrome.
Classical-like Ehlers-Danlos syndrome.
Vascular Ehlers-Danlos syndrome.
Cardiac-valvular Ehlers-Danlos syndrome.
Arthrochalasia Ehlers-Danlos syndrome.
Dermatosparaxis Ehlers-Danlos syndrome.
Kyphoscoliotic Ehlers-Danlos syndrome.
Brittle cornea syndrome.
Spondylodysplastic Ehlers-Danlos syndrome.
Musculocontractural Ehlers-Danlos syndrome.
Myopathic Ehlers-Danlos syndrome.
Periodontal Ehlers-Danlos syndrome.
They are classified by what they are like to experience and to examine (their presentation) and also by the gene changes that cause them.
Hypermobile Ehlers-Danlos syndrome
HEDS is the most common form of EDS. It is inherited but so far no gene has been identified as the cause. It can be mild and is probably often undiagnosed. However many people with hEDS can have significant pain and disability.
The main symptoms of hEDS are stretchy skin (which is not fragile but which may heal slowly) and unusually flexible joints.
Many people with hEDS have tiredness, pain and mood changes. The symptoms may include
Sleep disturbance and tiredness.
Fast heart rate.
Dizziness and fainting on standing up quickly.
Unexplained tummy pains, constipation and irritable bowel syndrome.
Tendency to nausea.
Anxiety and depression and panic attacks.
Problems with passing urine.
Widespread pain in the muscles and/or limbs.
Headaches.
Prolapse affecting the bladder, womb (uterus) or back passage.
How is hEDS diagnosed?
hEDS can be quite difficult to diagnose, as there is no specific test for it. As a result it often goes undiagnosed.
When symptoms are relatively mild, hEDS may be similar to benign joint hypermobility syndrome (BJHS). Some doctors think that the two conditions may be different degrees of the same thing.
Criterion 1: generalised joint hypermobility (GJH)
This is detected by doctors using a series of stretching and joint-bending tests which lead to the 'Beighton score'. GJH is diagnosed with a score or five or more (if aged under 50 years) and four or more (if aged over 50 years). Some doctors also use the 'five-point questionnaire' which asks :
Can you, or could you ever, put your hands flat on the floor without bending your knees?
Can you, or could you ever, bend your thumb to touch your forearm?
As a child, could you contort your body into strange shapes, or do the splits?
As a child or teenager, did your shoulder or kneecap dislocate more than once?
Do you consider yourself 'double-jointed'?
A 'yes' answer to two or more questions suggests likely hypermobile joints.
Criterion 2: two or more of the following (A-C) MUST be present
A: the presence of at least FIVE of the following:
Unusually soft or velvety skin.
Mild skin hyperextensibility.
Unexplained stretchmarks.
Piezogenic papules (little hard lumps under the skin) of the heel.
Recurrent or multiple abdominal hernia(s).
Thinned scarring in at least two places.
Any sort of prolapse without another reason, such as childbirth.
Tooth crowding and high, narrow palate.
Long, slender fingers (arachnodactyly).
Unusually long arms.
Floppy mitral (heart) valve.
Stretched aortic (heart) valve.
B: having a close family member diagnosed with hEDS.
C: having at least one of the following:
Pain in two or more limbs, daily for at least three months.
Widespread pain for ≥3 months.
Recurrent joint dislocations or instability.
Criterion 3: all the following MUST be the case
Skin must NOT be unusually fragile (this suggests other types of EDS).
No other medical explanation for symptoms.
Some of the common symptoms of hEDS, such as tiredness and tummy pain, are not included in the list of symptoms needed for diagnosis. This is because they are commonly found in many other conditions too, including other sorts of EDS, so they don't help identify hEDS specifically.
How is hEDS managed?
Regular gentle exercise and keeping your weight within the recommended range for your height will help fitness and stabilisation of joints with good, working muscles. Pilates can be beneficial in helping maintain core stability and maintaining good posture.
Some activities are better avoided, in order to protect the skin and joints from injury. Contact team sports, such as rugby and football, increase the risk of injury to joints, and it is better to choose less high-risk activities such as tennis or swimming.
Some people benefit from seeing specialists in pain management, rheumatology, physiotherapy or occupational therapy.
Painkillers may be helpful for generalised pains.
Some patients find talking therapies such as CBT helpful.
Is hEDS the same as joint hypermobility syndrome?
Some doctors believe that hEDS is a more severe form of hypermobility syndrome and there is certainly an overlap in symptoms. However, hypermobility syndrome mainly affects joints. EDS is more likely to be the diagnosis where there are associated, more serious conditions, such as heart valve problems, prolapse or repeated dislocations.
Classical Ehlers-Danlos syndrome
Classical EDS (cEDS) affects 1 in 20,000-50,000 people. It is autosomal dominant genetic condition, which means that inheriting one affected gene (one from either parent) causes the condition.
cEDS is similar to hEDS but in addition patients have very fragile skin, which is prone to splitting, particularly over the forehead, elbows, knees and chin. Those affected also have a tendency to form particularly thin scars. People also get rolled-up skin around joints which can be easily injured, little mobile nodules under the skin and easy bruising. Floppy muscles can be a feature, and children with cEDS may be slow to stand and walk.
cEDS is suspected if there is skin hyperextensibility and thin scars PLUS joint hypermobility. The other possible effects of cEDS include tiredness, poor sleep, tummy pains and prolapse. Sometimes there are problems with the heart valves, which can become floppy, causing a heart murmur.
Management of cEDS
The main way in which management of cEDS differs from hEDS is in the need to protect the fragile skin. The important things are:
Protecting the skin to minimise injuries with appropriate clothing and, when needed, protective pads and helmets.
Any injury that needs stitching should be done by a plastic surgeon. Stitches need to be left in place for longer than usual and wounds need extra support, with Steri-strips® or bandaging.
Children with cEDS need to find a balance between restrictions and risks. Contact sports such as rugby, ice hockey, boxing and martial arts, and high-impact activities like trampolining, are high-risk for injury and may need to be avoided. Lower-impact physical activities such as badminton, table tennis, bowling and swimming are better alternatives.
Regular gentle exercise is appropriate. Tiredness and joint pain can be part of cEDS and regular gentle exercise helps to reduce these effects.
Adults with cEDS may find Pilates helpful to build strength and help protect joints.
Physiotherapy is helpful when joint hypermobility is making joints slip out of place.
Occupational therapy can suggest aids to daily living and advise on pacing activities to avoid the extreme fatigue that can follow a period of over-activity.
There are some increased risks in pregnancy which are discussed in the separate section 'How do the Ehlers-Danlos syndromes affect pregnancy?', above.
Classical-like Ehlers-Danlos syndrome
This is also called tenascin-X deficient Ehlers-Danlos syndrome (clEDS).
This very rare condition is similar to cEDS, except that affected people do not make wide scars. It is autosomal recessive, which means that inheritance requires one faulty gene from EACH parent. If a parent has only one faulty gene each, they will generally NOT have the condition.
The other features of clEDS which are less common in other types of EDS are:
Long thin fingers.
'Flat' feet (pes planus).
Swollen legs.
Nerve compression syndromes (such as carpal tunnel syndrome).
Wasting of the muscles in the hands and feet.
Management of clEDS
clEDS is managed like cEDS, above. It is specifically recommended to avoid smoking.
Vascular Ehlers-Danlos syndrome
Vascular EDS (vEDS) is the most serious form of EDS. It affects about one in 50,000-200,000 people.
vEDS has autosomal dominant inheritance so just one copy of the defective gene from either parent will cause the condition. A person may also be affected if there is a new mutation in the gene, and so vEDS may occur in people with no history of the disorder in their family.
The main risk that people with vEDS face is leaking or bursting (rupture) of medium/large arteries. There is also a small risk of rupture of the large bowel (colon), and of the womb (uterus) during labour, and of spontaneous pneumothorax (caused by popping of an air space in the lungs). These are life-threatening effects, which usually occur in young adulthood, making this an extremely difficult diagnosis to be given.
Other features of vEDS include:
Early varicose veins.
Premature skin ageing on the hands and feet.
Fine, thinning hair.
Pointing and thinning of the cornea (the transparent front of the eye)
A characteristic facial appearance with small or absent earlobes, thin nose and lips and large eyes.
Hypermobile fingers and toes.
Management of vEDS
Treatment with a medicine called celiprolol greatly reduces the risk of arterial rupture. It may also be possible to repair blood vessels at risk of rupture before they do. This is a very specialised area.
vEDS patients should wear a medical alert bracelet, necklace or similar. The EDS National Diagnostic Service has produced a medic alert sheet with the information that may be needed in case of an emergency. It is best to avoid invasive tests or invasive treatments unless strictly necessary, because of the risk of damage to skin and blood vessels.
Cardiac-valvular Ehlers-Danlos syndrome
Cardiac-valvular EDS (cvEDS) causes problems with the heart valves, in addition to other, clEDS-like symptoms. It is a rare condition and inheritance requires one faulty gene from EACH parent. (If they only have one faulty gene each then they will generally NOT have cvEDS.)
Management is as for cEDS but with regular heart scans and monitoring, as the heart valves may need repair or replacement.
Arthrochalasia Ehlers-Danlos syndrome
Arthrochalasia EDS (aEDS) is a very rare, severe type of EDS in which the joints are very, very much looser than they should be. It has autosomal dominant inheritance so just one copy of the defective gene from either parent will cause the condition. It is usually diagnosed soon after birth, as babies are born with two dislocated hips. They also tend to have rather weak, floppy muscles, and particularly stretchy loose skin. Only around 30 cases have ever been described.
In addition to the severe joint problems, affected people also have fragile skin and, sometimes, a tendency to curvature of the spine.
Management of aEDS
The most important problem for children with aEDS is the early hip dislocation, since this severely affects mobility. Most of the usual surgical treatments for this condition do not work, so it is helpful if the diagnosis of aEDs is made before the hips are treated in order that a method that will succeed is chosen first.
Repeated dislocation of other joints can slow children from progress in standing and walking. The symptoms are worst in infancy and tend to improve with age so that some children manage to use orthoses (such as braces and callipers) to assist in walking later on.
Dermatosparaxis Ehlers-Danlos syndrome
Dermatosparaxis EDS (dEDS) is an extremely rare type of EDS which has been described only around ten times. Affected people have extremely fragile, sagging skin. It is usually diagnosed before the age of 2 years. Fragility, bruising and sagging of the skin are severe but, surprisingly, the skin heals well. Like the other rare types, inheritance requires one faulty gene from EACH parent. If a parent has only one faulty gene, they will generally NOT have the condition.
Affected children tend to have characteristic facial features, such as prominent eyes with blue or grey sclerae (the whites of the eyes) and a small chin. They often have other features seen in classical EDS, such as:
Hypermobile joints.
Hernias.
Tiredness.
'Thinning' of the bones.
Floppy muscles.
Constipation.
Tummy pain.
They can also experience abnormalities of the teeth, gums and vision, requiring spectacles.
Management of dEDS
Management is as for cEDS. A specialist dentist will need to be involved if there are tooth and gum problems, and an ophthalmologist if there are eye problems.
Kyphoscoliotic Ehlers-Danlos syndrome
This is a rare type of EDS which has been described only around 60 times. Like the other rare types, in order to inherit kyphoscoliotic EDS (kEDS), inheritance requires one faulty gene from each parent. If a parent has only one faulty gene each, they will generally NOT have the condition. It causes worsening (progressive) curvature of the spine (kyphoscoliosis).
Babies with kEDS are very floppy and they stand and walk late. Eyes are also affected and may have a thin surface (sclera) which appears slightly blue and which can (rarely) rupture. Some affected children have hearing problems. Other signs of this type of EDS include thin bones and long thin fingers
Joints are very hypermobile and there may be frequent dislocations. Skin may be stretchy, soft and fragile, bruise easily and form widened, atrophic scars.
Management of kEDS
This is as for classical EDS but with particular focus on trying to keep the back growing straight and involving a specialist optician to look after the eyes.
Brittle cornea syndrome
In this rare type of EDS the front coverings of the eye are thinner and less strong than usual and the white of the eye (sclera) may appear blue. Like the other rare types, inheritance requires one faulty gene from each parent. If a parent has only one faulty gene, they will generally NOT have the condition.
People with brittle cornea syndrome are generally very short-sighted and the eye itself is at risk of rupturing. They also have the typical skin and joint features of classical and hypermobility types of EDS.
Management of brittle cornea syndrome
This is as for cEDS, with additional care needed for the eyes.
Spondylodysplastic Ehlers-Danlos syndrome
Spondylodysplastic EDS (spEDS) is a severe EDS. Like the other rare types, inheritance requires one faulty gene from each parent. If a parent has only one faulty gene each, they will generally NOT have the condition. It causes short stature and weak floppy muscles. Bowing of limbs is common.
Unlike other types, there may be learning difficulties in children. Some of the features of EDS affecting eyes, bones, joints, blood vessels or the lungs may be present and there is a characteristic facial appearance.
Management of spEDS
Management involves the approaches used in cEDS towards skin and joints, together with specialist physiotherapy as the child grows up - to help maximise their bone growth and strength. It is not possible to generalise about the degree of learning difficulty that children experience as the condition is so rare.
Musculocontractural Ehlers-Danlos syndrome
Musculocontractural EDS (mcEDS) is a rare type of EDS in which muscles in the legs and arms are severely tightened and shortened - these are called muscle contractures. The skin is severely affected too, being very stretchy, fragile and easily bruised, with the atrophic scars seen in clEDS. It can also affect bones, eyes, the digestive system and the kidneys. Like the other rare types, inheritance requires one faulty gene from each parent. If a parent has only one faulty gene, they will generally NOT have the condition.
Management of mcEDS
Management of this condition follows the patterns detailed above for cEDS, with special attention to any of the eye, bone, kidney or digestive symptoms.
Myopathic Ehlers-Danlos syndrome
Myopathic EDS (mEDS) is a milder subtype of EDS which causes very floppy muscles which may seem very poorly developed at birth but which improve with age. It is possible to inherit mEDS if just one parent is affected although some 'versions' of mEDS seem to require inheritance of a faulty gene from both parents.
Management of mEDS
Management is as for the other types, with a focus on physiotherapy and muscle strengthening.
Periodontal Ehlers-Danlos syndrome
Periodontal EDS (pEDS) causes dental problems, particularly inflamed or absent gums and poor tooth development, on top of typical features of cEDS. It is rare but only one faulty gene (from either parent) is needed to develop the condition.
Management of pEDS
Management of pEDS is as for management of hEDS and cEDS but with additional, specialist dental care.
Summary
EDS is a very varied condition, which primarily affects skin and joints but which can also affect many other organs. Thirteen subtypes have now been classified.
There is no specific cure for hEDS but if symptoms are interfering with life, then physiotherapy, gentle exercise, talking therapies and, sometimes, pain relief are the mainstays of treatment. With the other types of EDS, specialist teams should be involved in the care, depending on which organs are affected.
Dr Mary Lowth is an author or the original author of this leaflet.
Patient picks for Genetic conditions
Children's health
Cystic fibrosis
Cystic fibrosis is an inherited disease which mainly affects the lungs and pancreas but can involve other organs. Symptoms usually begin in early childhood and include persistent cough, wheeze, repeated chest infections, difficulty digesting food and general ill health. Treatments include antibiotics, physiotherapy, medicines for thinning mucus, pancreatic enzyme replacements and other therapies.
by Dr Toni Hazell, FRCGP

Children's health
Wilson's disease
Wilson's disease is a genetic disorder in which copper builds up in the body, mainly in the liver and brain. Without any treatment, the build-up of copper can cause serious symptoms. Treatment is with medication to remove the excess copper and/or to prevent a further build-up of copper.
by Dr Philippa Vincent, MRCGP
Frequently asked questions
Can Ehlers-Danlos syndromes (EDS) shorten a person's life expectancy?
The outlook for EDS varies depending on the type. Most forms of EDS do not affect a person's lifespan, and those with milder symptoms can live without significant restrictions. However, for individuals with the rare vascular type of EDS, there is a serious risk of blood vessel or internal organ rupture, which can lead to premature death.
Are there different patterns of inheritance for the Ehlers-Danlos syndromes?
Yes, EDS can be inherited in different ways. Some types, like hypermobile, classical, and vascular EDS, follow an autosomal dominant inheritance pattern, meaning a child has a 1 in 2 chance of being affected if one parent has the condition. Other types, such as kyphoscoliotic EDS, are inherited in an autosomal recessive manner, where both parents must contribute a faulty gene for the child to be affected, resulting in a 1 in 4 chance. Occasionally, EDS can also be caused by a new, random gene mutation rather than being inherited.
Can EDS be mistaken for other conditions?
Yes, some milder forms of EDS, particularly hypermobile EDS (hEDS), can be mistaken for conditions like chronic fatigue syndrome or fibromyalgia due to overlapping symptoms such as tiredness, widespread pain, and sleep disturbances.
What kind of exercises are recommended for people with EDS?
Exercise is important for people with EDS, but it should be chosen to minimise the risk of injury. Good options include swimming, walking, tai chi, or using a stationary bike. Regular gentle exercise helps build strength, improve balance, and support joints by strengthening muscles. Activities with a high risk of physical injury, such as contact sports (e.g., rugby, football) or high-impact activities (e.g., trampolining), should ideally be avoided.
Is it possible to have Ehlers-Danlos syndromes without knowing it?
Yes, milder forms of EDS are often not diagnosed until early adulthood because it can take some time before the symptoms and signs become noticeable. For example, hypermobile EDS (hEDS), the most common type, can be mild and often goes undiagnosed.
Further reading and references
- Syx D, De Wandele I, Rombaut L, et al; Hypermobility, the Ehlers-Danlos syndromes and chronic pain. Clin Exp Rheumatol. 2017 Sep-Oct;35 Suppl 107(5):116-122. Epub 2017 Sep 28.
- Gensemer C, Burks R, Kautz S, et al; Hypermobile Ehlers-Danlos syndromes: Complex phenotypes, challenging diagnoses, and poorly understood causes. Dev Dyn. 2021 Mar;250(3):318-344. doi: 10.1002/dvdy.220. Epub 2020 Aug 17.
- Islam M, Chang C, Gershwin ME; Ehlers-Danlos Syndrome: Immunologic contrasts and connective tissue comparisons. J Transl Autoimmun. 2020 Dec 20;4:100077. doi: 10.1016/j.jtauto.2020.100077. eCollection 2021.
- Bascom R, Dhingra R, Francomano CA; Respiratory manifestations in the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2021 Dec;187(4):533-548. doi: 10.1002/ajmg.c.31953. Epub 2021 Nov 22.
- Ishiguro H, Yagasaki H, Horiuchi Y; Ehlers-Danlos Syndrome in the Field of Psychiatry: A Review. Front Psychiatry. 2022 Jan 11;12:803898. doi: 10.3389/fpsyt.2021.803898. eCollection 2021.
About the authorView full bio

Dr Rachel Hudson, MRCGP
General Practitioner and Medical Author
MBChB, MRCGP (2008), BSc (Medical Science), DFSRH, DRCOG, DCH
Dr Rachel Hudson, is an NHS GP working in the North West of England.
About the reviewerView full bio

Dr Colin Tidy, MRCGP
General Practitioner, Medical Author
MBBS, MRCGP, MRCP (Paediatrics), DCH
Dr Colin Tidy is an NHS Doctor, based in Oxfordshire.
Article history
The information on this page is written and peer reviewed by qualified clinicians.
Article also available in English, German, Spanish, French, Italian, Portuguese, Hindi, Hebrew, Arabic, and Swedish.
Next review due: 7 Oct 2027
8 Oct 2024 | Latest version

Ask, share, connect.
Browse discussions, ask questions, and share experiences across hundreds of health topics.

Feeling unwell?
Assess your symptoms online for free
Sign up to the Patient newsletter
Your weekly dose of clear, trustworthy health advice - written to help you feel informed, confident and in control.
By subscribing you accept our Privacy Policy. You can unsubscribe at any time. We never sell your data.
More in children's health
- Back pain in children
- BCG immunisation
- Bedwetting medicine - desmopressin
- Bullying
- Cerebral palsy
- Childhood gastro-oesophageal reflux
- Childhood obesity
- Cot death
- Depression in children
- Feeding your baby
- Glue ear
- Head lice and nits
- Knock knees
- Mumps
- Neonatal jaundice
- Perthes' disease
- Pneumococcal immunisation
- Retinopathy of prematurity
- Treating newborn health problems
- Wilms' tumour