Bullous pemphigoid
Peer reviewed by Dr Doug McKechnie, MRCGPLast updated by Dr Philippa Vincent, MRCGPLast updated 6 Aug 2025
Meets Patient’s editorial guidelines
- DownloadDownload
- Share
- Language
- Discussion
- Audio Version
- Add to preferred sources on Google
Medical Professionals
Professional Reference articles are designed for health professionals to use. They are written by UK doctors and based on research evidence, UK and European Guidelines. You may find the Bullous pemphigoid article more useful, or one of our other health articles.
What is bullous pemphigoid?
Bullous pemphigoid (BP) is the most common autoimmune subepidermal blistering disease, affecting predominantly elderly people (blistering is intraepidermal in pemphigus).
It is a chronic condition, characterised by generalised pruritic urticarial plaques and tense subepithelial blisters.1
How does bullous pemphigoid develop? (pathogenesis)
An abnormal T-cell immune response and synthesis of IgG and IgE autoantibodies against hemidesmosomal proteins (BP180 and BP230) lead to neutrophil chemotaxis and degradation of the basement membrane zone.2
The pathogenesis depends on the interaction between predisposing factors, such as human leukocyte antigen (HLA) genes, comorbidities, ageing, and trigger factors. Several trigger factors, such as drugs, thermal or electrical burns, surgical procedures, trauma, ultraviolet irradiation, radiotherapy, chemical preparations, transplants and infections, may induce or exacerbate bullous pemphigoid.1
There is a significant association between bullous pemphigoid and psoriasis.3
How common is bullous pemphigoid? (Epidemiology)4
BP is the most common autoimmune blistering disease.
BP usually occurs in individuals over the age of 60, with a median age at presentation in the UK of 80 years.
In the UK, the incidence is 46 per 100,000 persons per year among those over 90 years old, but only 1.5 per 100,000 among those 50-59 years old.
It has been typically thought to affect men and women equally with no racial bias5 but some studies suggest that Black people have greater disease activity whilst others suggest it is more common in the Jewish population.3
It has very occasionally been reported in children.5
Symptoms of bullous pemphigoid (presentation)6
Bullous pemphigoid classically presents with tense blisters over urticarial plaques on the trunk and extremities accompanied by intense pruritus.2
The initial presentation of pemphigoid is very variable, as blistering skin lesions may only occur late in the course of the disease.
Symptoms and signs may present with a subacute or acute onset. Symptoms and signs which may be seen in patients with all forms of bullous pemphigoid include:
Itch, or pruritus, which is frequently a feature. This may occur weeks or months before the appearance of any visible skin lesion.
Rash. An urticarial or erythematous rash may precede the appearance of the blisters.
Blisters or bullae, which are typical and often occur in skin flexures. Depending on the form of bullous pemphigoid these may affect a single site or be more widespread.
Mucous membranes. Blisters appear mainly on palatal mucosa. They may be clinically insignificant but can cause problems with dysphagia.
Different clinical forms
There are several distinct clinical presentations.
Generalised bullous
This is the most common form:
Bullous pemphigoid leg blisters
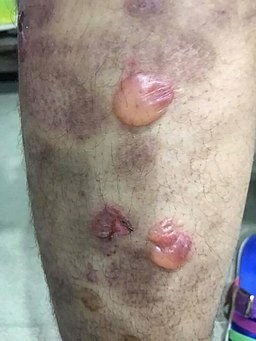
© Mohammad2018, CC BY-SA 4.0, via Wikimedia Commons
Tense bullae can form anywhere but commonly around flexural areas. They can appear both on normal skin and on erythematous skin.
Mucosal involvement is rare and not clinically significant when it does occur.
The bullae heal without scarring.
Vesicular
This is a less common form:
It presents typically with small groups of tense blisters which collect on erythematous areas of skin (rather than normal skin).
Mucous membrane pemphigoid includes a group of autoimmune bullous diseases characterised by subepithelial blisters, erosions and scarring of mucous membranes, skin or both.7
Other rarer forms include vegetative, generalised erythroderma, urticarial, nodular (or pemphigoid nodularis) and acral (a childhood form associated with vaccination).8
Differential diagnosis9
The differential diagnoses will include:
Cicatricial pemphigoid (this is a complex condition where skin lesions are usually trivial and mucosal lesions heal with scarring. Several different autoantibodies have been implicated).
Other conditions which may be considered include:
Other blistering skin disorders such as chickenpox.
Impetigo, which may be mistaken for the various stages of BP.
Urticaria.
The bullous eruption of systemic lupus erythematosus.
Herpes gestationis or gestational pemphigoid. This presents in the late second or third trimester, and resolves on delivery. It is immunologically identical to BP.
Diagnosing bullous pemphigoid (investigations)10
Diagnosis is based on clinical manifestations and confirmed with histological, immunofluorescence, and serological testing. Serological detection of the autoantibodies largely relies on indirect immunofluorescence (IIF) and enzyme-linked immunosorbent assay (ELISA) tests.
Direct immunofluorescence (DIF) studies.11 DIF demonstrates IgG in up to 90% of patients and C3 deposition in nearly 100% of patients. DIF can differentiate BP from cicatricial pemphigoid and epidermolysis bullosa acquisita by incubating the skin biopsy sample in a salt solution before DIF.
The best skin samples for diagnosis come from apparently normal skin near a bullous pemphigoid lesion.12 Fresh specimens are best, as transport media may give false negative results.
Histological tests. These reveal subepidermal blisters with inflammatory infiltrate and usually eosinophilic predominance.
Associated diseases
It had been thought that bullous pemphigoid may be associated with the presence of malignant tumours; however, age- and sex-matched studies have concluded that no such association exists.
Bullous pemphigoid treatment10 13
The effectiveness of topical therapy with potent topical corticosteroids in localised and moderate forms of bullous pemphigoid is supported by a Cochrane review.14
In localised, non-severe forms, immunomodulatory, non-immunosuppressant drugs such as doxycycline may be considered. Although its exact mechanism of action is not well understood, doxycycline proved to be as effective as oral prednisolone for short-term blister control and is safer in the long term.
Systemic corticosteroid therapy (prednisone 0.5 mg/kg/day) is the treatment of choice for severe forms. For maintenance treatment, doses may be tapered gradually within 4-6 months of initiation of treatment.
Adjunctive therapy includes combination with azathioprine, mycophenolate mofetil, tetracyclines plus nicotinamide, methotrexate and dapsone.
Available data suggest that rituximab may provide clinical benefits for patients with refractory bullous pemphigoid.
Bacterial superinfection of erosions should be treated with local antiseptics.
Wound dressings should be considered for large wounds. Sterile puncture of large blisters is recommended.
Complications of bullous pemphigoid
Secondary infections.
Immunosuppression.
Corticosteroid side-effects (hypertension, diabetes, osteoporosis, heart disease).
Prognosis
Bullous pemphigoid is a chronic inflammatory disease which can persist with remissions and exacerbations for months or years.
It typically resolves in a few months but can remain for up to 5 years.5
There is twice as high a risk of death compared with age-matched controls.34
Elderly patients are also particularly at risk from the immunosuppressive effects of therapy, which may place them at greater risk of life-threatening events than the disease itself.
Patients on corticosteroids and immunosuppressants are at risk of serious side-effects - for example, peptic ulcer disease, agranulocytosis and diabetes.
Patients with bullous pemphigoid have been shown to have a high prevalence of neurological comorbidities, including dementia, stroke, Parkinson's disease and multiple sclerosis.3
Exclusive updates for healthcare professionals
Stay informed with the latest clinical updates, professional insights, and evidence-based guidance. The Patient Pro newsletter curates essential content for healthcare professionals—delivered straight to your inbox.
By subscribing you accept our Privacy Policy. You can unsubscribe at any time. We never sell your data.
Further reading and references
- Moro F, Fania L, Sinagra JLM, et al; Bullous Pemphigoid: Trigger and Predisposing Factors. Biomolecules. 2020 Oct 10;10(10). pii: biom10101432. doi: 10.3390/biom10101432.
- Miyamoto D, Santi CG, Aoki V, et al; Bullous pemphigoid. An Bras Dermatol. 2019 Mar-Apr;94(2):133-146. doi: 10.1590/abd1806-4841.20199007. Epub 2019 May 9.
- Bullous pemphigoid burden of disease, management and unmet therapeutic needs; V P Werth et al
- Hammers CM, Stanley JR; Mechanisms of Disease: Pemphigus and Bullous Pemphigoid. Annu Rev Pathol. 2016 May 23;11:175-97. doi: 10.1146/annurev-pathol-012615-044313. Epub 2016 Feb 22.
- Baigrie D, Nookala V; Bullous Pemphigoid.
- Khandpur S, Verma P; Bullous pemphigoid. Indian J Dermatol Venereol Leprol. 2011 Jul-Aug;77(4):450-5. doi: 10.4103/0378-6323.82398.
- Murrell DF, Marinovic B, Caux F, et al; Definitions and outcome measures for mucous membrane pemphigoid: Recommendations of an international panel of experts. J Am Acad Dermatol. 2015 Jan;72(1):168-174. doi: 10.1016/j.jaad.2014.08.024. Epub 2014 Nov 4.
- de la Fuente S, Hernandez-Martin A, de Lucas R, et al; Postvaccination bullous pemphigoid in infancy: report of three new cases and literature review. Pediatr Dermatol. 2013 Nov-Dec;30(6):741-4. doi: 10.1111/pde.12231. Epub 2013 Oct 11.
- Damoiseaux J; Bullous skin diseases: classical types of autoimmune diseases. Scientifica (Cairo). 2013;2013:457982. doi: 10.1155/2013/457982. Epub 2013 Jan 8.
- Di Lernia V, Casanova DM, Goldust M, et al; Pemphigus Vulgaris and Bullous Pemphigoid: Update on Diagnosis and Treatment. Dermatol Pract Concept. 2020 Jun 29;10(3):e2020050. doi: 10.5826/dpc.1003a50. eCollection 2020 Jul.
- Schmidt E, della Torre R, Borradori L; Clinical features and practical diagnosis of bullous pemphigoid. Dermatol Clin. 2011 Jul;29(3):427-38, viii-ix.
- Guidelines for the management of bullous pemphigoid; British Association of Dermatologists (2012)
- Yamagami J; Recent advances in the understanding and treatment of pemphigus and pemphigoid. F1000Res. 2018 Aug 30;7. doi: 10.12688/f1000research.14474.1. eCollection 2018.
- Kirtschig G, Middleton P, Bennett C, et al; Interventions for bullous pemphigoid. Cochrane Database Syst Rev. 2010 Oct 6;(10):CD002292.
About the authorView full bio

Dr Philippa Vincent, MRCGP
General Practitioner, Medical Author
MB BS, Bsc, MRCGP (2000), DCH, DFSRH, DRCOG
Dr Philippa Vincent is an NHS GP working in North London.
About the reviewerView full bio

Dr Doug McKechnie, MRCGP
Medical Writer
MA, MBBS, MSc, DRCOG, MRCP(UK), MRCGP(2021), FHEA
Dr Doug McKechnie is an NHS GP working in London. He works full-time clinically and is also the Deputy Lead for the Clinical and Professional Practice module at University College London Medical School.
Article history
The information on this page is written and peer reviewed by qualified clinicians.
Article also available in English, German, Spanish, French, Italian, Portuguese, Hindi, Hebrew, Arabic, and Swedish.
Next review due: 5 Aug 2028
6 Aug 2025 | Latest version

Ask, share, connect.
Browse discussions, ask questions, and share experiences across hundreds of health topics.

Feeling unwell?
Assess your symptoms online for free
More in dermatology
- Acne conglobata and rarer forms of acne
- Atopic dermatitis and eczema
- Becker's naevus
- Bowen's disease
- Buruli ulcer
- Candidiasis
- Chondrodermatitis nodularis
- Dermatological history and examination
- Erythema annulare centrifugum
- Febrile neutrophilic dermatosis
- Henoch-Schönlein purpura
- Hidradenitis suppurativa
- Hirsutism
- Lichen planus
- Lichen sclerosus
- Living with skin disease
- Pathogenic free-living amoeba
- Pemphigoid gestationis
- Smallpox
- Vasculitis