Episcleritis and scleritis
Peer reviewed by Dr Colin Tidy, MRCGPLast updated by Dr Hayley Willacy, FRCGP Last updated 17 Jun 2025
Meets Patient’s editorial guidelines
- DownloadDownload
- Share
- Language
- Discussion
- Audio Version
Medical Professionals
Professional Reference articles are designed for health professionals to use. They are written by UK doctors and based on research evidence, UK and European Guidelines. You may find the Episcleritis and scleritis article more useful, or one of our other health articles.
In this article:
Continue reading below
Background
Episcleritis and scleritis are separate conditions which can present with some similar features but differ dramatically in significance. Episcleritis does not progress to scleritis.
Episcleritis is inflammation of the superficial, episcleral layer of the eye. It is relatively common, benign and self-limiting.
Scleritis is inflammation involving the sclera. It is a severe ocular inflammation, often with ocular complications, which nearly always requires systemic treatment.1 2
Differentiating episcleritis from scleritis requires careful examination and an understanding of the anatomy. The blood vessels of the episclera are not easily seen in the non-inflamed eye. There are three plexuses.3
Bulbar conjunctival plexus - a superficial plexus of fine vessels overlying and freely moveable over the episclera. Mainly arteries, when inflamed they are bright red.
Episcleral plexus - straight radially arranged vessels in the superficial episclera. Mainly veins, they are also moveable over the deep layers (although less easily than the bulbar conjunctival vessels). When inflamed they give the eye a salmon pink colour.
Deep episcleral plexus (also called scleral plexus) - a criss-cross of vessels closely applied to the sclera. When inflamed this layer looks bluish red and is immobile.
Examination can determine the depth of inflammation in the eye by observing which of the layers of blood vessels are inflamed.
Episcleritis
Back to contentsEpiscleritis involves inflammation of the episclera. There are generally considered to be two types:4
Simple episcleritis: characterised by vascular congestion on an even episcleral surface. This can affect a single segment of the episclera or all of it (diffuse episcleritis).
Nodular episcleritis: characterised by a discrete elevated area of inflamed episclera.
Episcleritis
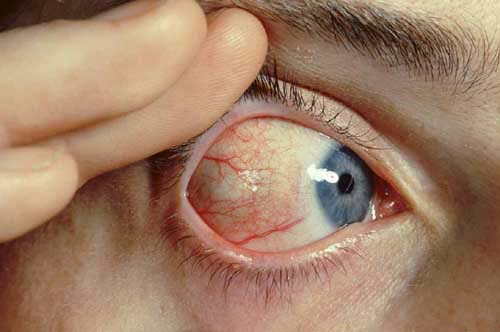
© Asagan, CC BY-SA 3.0, via Wikimedia Commons
By Asagan, CC BY-SA 3.0, via Wikimedia Commons
Episcleritis lasts 7-10 days before spontaneously resolving. If there are underlying systemic predisposing conditions, episodes may be more prolonged.
Nodular episcleritis is more severe and takes longer to resolve. It is also much more likely to be associated with systemic disease.
Continue reading below
Scleritis4
Back to contentsThis is much more severe inflammation that occurs throughout the entire thickness of the sclera.
Scleritis eye inflammation
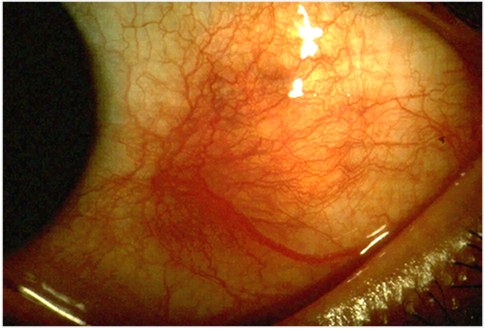
© Kribz, CC BY-SA 3.0, via Wikimedia Commons
By Kribz, CC BY-SA 3.0, via Wikimedia Commons
Scleritis is rare. It occurs more frequently in women and generally in an older age group than patients with episcleritis, with an age range of 40-60 years.
It is traditionally classified according to which part of the sclera is affected, a classification developed in 1976.3 It may be anterior (90% of cases) or posterior (10% of cases) and there are four clearly recognisable groups of the anterior form:
Anterior scleritis (90% of cases):
Diffuse anterior - this is the most common (and benign) form, characterised by widespread inflammation of the anterior sclera. It accounts for about 50% of scleritis cases. It is not usually sight-threatening and tends to resolve.
Nodular - there are erythematous, tender, fixed nodules in the sclera which may (1 in 4 cases) progress to necrotising scleritis. It commonly recurs.
Necrotising - this is less frequent, accounting for around 15% of cases and is characterised by extreme pain and marked scleral damage; it is usually associated with underlying systemic disease. It is divided into:
Necrotising scleritis with corneal inflammation. This is also known as sclerokeratitis.
Necrotising scleritis without inflammation (scleromalacia perforans) - (4% of cases of scleritis). This is notable for its lack of symptoms. It is a severe disorder of the globe with insidious onset, slow progression and a lack of symptoms until the bare choroid is seen under the thin layer of the conjunctiva. It is bilateral and is only seen with advanced rheumatoid arthritis (usually in women).
Posterior scleritis (10% of cases) - affects the back of the eye and is rare and difficult to diagnose. It affects women more often than men. It can present with severe eye pain, retinal detachment, choroidal folds and loss of vision. About one third of patients with posterior scleritis have associated anterior scleritis. Posterior scleritis can lead to rapid permanent visual loss.
The diffuse and nodular types are the most prevalent, accounting for around 90%.
Particularly important is the recognition of necrotising scleritis, which is frequently associated with greater complications and worse outcomes. Necrotising scleritis needs early, aggressive therapy, as conventional steroid therapy is generally insufficient.
The severity of scleritis is typically graded on a scale of 1-4 as there is no standardised system for grading severity. Standardisation of this grading would be useful both for patient care and for research. Various systems which include area and severity of inflammation have been proposed but not universally adopted.5 6
Epidemiology
Back to contentsEpiscleritis
Most cases of episcleritis have no identifiable cause, although a small number are associated with systemic inflammatory conditions.
The condition can be recurrent, with attacks typically recurring every few months.
The prevalence is uncertain (as many patients probably do not present) but it occurs most frequently in younger patients: young adult to fifth decade.
A 2013 study estimated incidence of episcleritis as 41.0 per 100,000 per year and prevalence at 52.6.7
Scleritis
Scleritis often appears in association with other inflammatory diseases such as rheumatoid arthritis and granulomatosis - the histopathological changes are characteristic of a chronic granulomatous disease.
Scleritis may be the first symptom of onset of connective tissue disease.
A 2013 study estimated the incidence of scleritis at 3.4 per 100,000 per year and the prevalence at 5.2.7
Continue reading below
Presentation4
Back to contentsIt is important to distinguish between the two conditions in a systematic manner and specifically to rule out scleritis when diagnosing episcleritis.
Both conditions present with a red eye which may be painful.
Episcleral vessels can be moved with a cotton bud. When phenylephrine 10% is applied, they blanch (this will also dilate the pupil).
Scleral vessels appear darker, follow a radial pattern, are immobile and do not blanch.
Episcleritis is generally a mild condition with no associated ocular symptoms; scleritis tends to cause severe pain with associated symptoms. Occasionally, however, the symptoms of scleritis can be mild, particularly early in the condition (which can have a gradual onset).
Scleritis is best detected by examining the sclera in daylight, retracting the lids to obtain the best view.
Visual acuity testing will usually be normal in episcleritis but can be normal in scleritis.
Slit-lamp examination will detect intraocular inflammation in scleritis and may help assess severity. CT scan, MRI and ultrasound may also help determine extent of involvement.
The table below compares and contrasts the signs and symptoms of typical cases.
| Episcleritis | Scleritis |
Presentation | Acute onset of redness with discomfort (more gradual in nodular episcleritis). May present late, as symptoms mild. Discomfort, grittiness, aching in or around the eye. Very rarely, marked pain. 40% bilateral. Localised or diffuse red eye. Watering and occasional mild photophobia. No other associated ocular symptoms. No discharge other than watering. Visual acuity normal. | Subacute or gradual onset (In scleromalacia perforans and posterior scleritis there may be no anterior redness). Presents early, as symptoms are severe. Boring eye pain, often radiating to the forehead, brow and jaw and usually severe. Worst in necrotising scleritis; may be mild or absent in scleromalacia perforans. Pain worse with movement of the eye and at night (may wake the patient). 50% bilateral. Localised or diffuse red eye. Associated watering, photophobia. No discharge other than watering. Gradual decrease in vision. Diplopia (posterior disease). Occasional associated systemic symptoms (fever, vomiting, headache). Scleromalacia perforans may present with minimal/no symptoms. Posterior scleritis may present with very severe symptoms but a quiet white eye. Rarely, posterior scleritis presents with decreased visual acuity in the absence of pain.3 8 |
Past history | Recurrent episodes are common. May be associated with systemic disease but this is unusual. | Recurrent episodes are common. Commonly associated with systemic disease which may be severe. |
Signs relating to presentation | Sectoral/diffuse redness. Engorged episcleral vessels extending radially. Translucent white nodule may be present within the inflamed area. Visual acuity is normal. | Visual acuity may be reduced or normal. Anterior Sectoral or diffuse redness. Scleral, episcleral and conjunctival vessels all involved. Sclera may take on a bluish tinge (best seen on gross examination in natural daylight) ± may be thin and oedematous. The globe is tender. Scleromalacia perforans: may be enlarging and coalescing yellow necrotic nodules ± scleral thinning (actual perforation is rare unless intraocular pressure is elevated). Posterior Lid oedema. Proptosis. Optic disc swelling. Retinal detachment can occur. |
Other ocular signs | Corneal involvement is possible but rare. Up to 10% of cases have an associated anterior uveitis. | May be associated with: Corneal involvement. Uveitis. Cataract formation. Rapid-onset refractive changes. |
Differential diagnosis
Back to contentsEpiscleritis/scleritis (each is in the differential diagnosis list for the other).
Posterior scleritis has a more extensive list of differential diagnoses including:
Orbital inflammatory disease/mass.
Uveal effusion syndrome.
Vogt-Koyanagi-Harada disease, a granulomatous uveitis with retinal detachment mainly seen in Asian people, secondary to tuberculosis or toxoplasmosis.9
Intraocular lymphoma.
Episcleritis - investigations
Back to contentsA simple case of episcleritis need not be investigated once a thorough history is taken and examination is carried out to rule out the possibility of an associated systemic disease (see 'Associated diseases', below). If there is a suspicion, biochemical tests should be performed as guided by concern (for example, antinuclear antibody, rheumatoid factor, serum uric acid levels, etc). Patients who are more likely to have associated conditions include those with nodular disease and individuals with severe recurrent episodes.
Episcleritis - associated diseases10
Back to contentsThis condition is most commonly idiopathic but 30% of cases are associated with underlying systemic disease.
The most commonly associated systemic disorders are ulcerative colitis and Crohn's disease.
Hyperuricaemia is seen in about 11%.
Connective tissue disease such as rheumatoid arthritis, polyarteritis nodosa, systemic lupus erythematosus and granulomatosis with polyangiitis (GPA) have also been associated with recurring episcleritis.
Infectious causes are less common but include herpes zoster virus, herpes simplex virus, Lyme disease, syphilis, hepatitis B and brucellosis.1112 More rarely, fungi and parasitic infestations may be associated with episcleritis.
Miscellaneous associations include rosacea, atopy, lymphoma and thyroid eye disease.
A foreign body or chemical insult may also cause episcleritis.
There are some very rare but documented associations with a number of other conditions, including T-cell leukaemia, paraproteinaemia, paraneoplastic syndromes, Wiskott-Aldrich syndrome, adrenal cortical insufficiency and progressive hemifacial atrophy.
Episcleritis - management10
Back to contentsEpiscleritis can be uncomfortable, although it is not usually described as painful by patients. and treatment focuses on decreasing symptoms with lubricating eye drops of ointment. More severe cases may be treated with topical corticosteroids or oral anti-inflammatory medications.
Artificial tears may provide some relief, particularly in nodular disease.
The use of topical non-steroidal anti-inflammatory drugs (NSAIDs) can be helpful in more symptomatic patients.
Where the episode is more severe, a short course of topical steroids may be required (under the supervision of an ophthalmologist).
Nodular episcleritis may respond best to oral NSAIDs.
It is important to review patients (a week after initial presentation is adequate) to ensure resolution of symptoms.
If the episode is severe, not resolving or recurs more than three times, it is appropriate to refer to the Eye Clinic.
Episcleritis - complications4
Back to contentsInvolvement of ocular structures is rare: there may occasionally be corneal involvement (in the form of inflammatory cell infiltrates or oedema) and recurrent attacks over a period of years may induce some degree of slight scleral thinning. Complications more commonly occur in those patients who have repeatedly required topical steroid treatment over a number of years. They include cataract formation, ocular hypertension and steroid-induced glaucoma. There may also be rebound inflammation from steroid withdrawal.
Episcleritis - prognosis10
Back to contentsThis is a mild, self-limiting disease which tends to resolve fully over 1-2 weeks (5-6 weeks in nodular episcleritis) but patients should be aware that episodes may recur in the same or the fellow eye. Treatment-related complications are the most common cause of visual impairment in these patients (hence the need to recognise the benign nature of this disease and not to overtreat).
Scleritis - investigations
Back to contentsIn cases of scleritis, frequency of association and the severity of these associated problems means that it has to be assumed that there is an underlying cause until proven otherwise. Every effort should be made to rule out associated pathology.
Depending on clinical suspicion, biochemical tests (for example, FBC and inflammatory markers, rheumatoid screen, syphilis screen), urine dipstick for blood and protein, B-scan ultrasonography of the globe, plain X-rays (chest and sacroiliac joints), MRI or CT imaging (sinuses and orbits) may be indicated.
In a patient with no previously diagnosed systemic disease, it is particularly important to rule out systemic vasculitis, as this is the least likely to have been previously diagnosed and it is a potentially life-threatening disorder.13
Usually, investigations will be carried out jointly between the ophthalmologists and other relevant hospital specialists.
Scleritis - associated diseases14
Back to contentsAt least 50% of patients have an underlying autoimmune condition.
In 15% of cases, the scleritis is the first presentation of a collagen vascular disorder, preceding it by one to several months.15
Scleritis is frequently associated with connective tissue diseases of which rheumatoid arthritis is by far the most common. Other connective tissue diseases seen are GPA, relapsing polychondritis, systemic lupus erythematosus, reactive arthritis, polyarteritis nodosa and ankylosing spondylitis.
Other common associations include gout, eosinophilic granulomatosis with polyangiitis and syphilis.11
Scleritis may occur following ocular surgery. It tends to present as intense inflammation adjacent to the surgical site, usually within six months of the procedure. There is an association with the presence of underlying disease.16
Less common associations include tuberculosis and local infections (usually spreading from a corneal ulcer) - common causative organisms are Pseudomonas aeruginosa, Streptococcus pneumoniae, Staphylococcus aureus and varicella-zoster virus. It may also occur with Lyme disease. Fungal scleritis is very rare but should be suspected when the condition is not responding to immunosuppressive therapy.17
More unusual causes of scleritis include:
Scleritis - management2122
Back to contentsSuspect scleritis should be referred immediately to the ophthalmologists. Patients should all be under specialist care (usually co-managed by the ophthalmologists and rheumatologists).
Management will depend on the type of scleritis and on whether there is any underlying systemic disease.
All patients with scleritis should have any underlying disease addressed urgently.
In cases of infectious scleritis, systemic antibiotics will always be required and immunosuppressants must be used with particular care.
Diffuse anterior scleritis and nodular scleritis
This is treated initially with oral NSAIDs (for example, ibuprofen 400 mg qds).
If oral NSAIDs are not effective then oral prednisolone (for example, 80 mg od) can be used.
A subconjunctival or periorbital steroid injection may be effective although this remains controversial.
If this is not effective, immunosuppressive therapy such as methotrexate, azathioprine, mycophenolate mofetil, cyclophosphamide, or ciclosporin may be helpful. It may take several weeks to adjust the treatment to produce a satisfactory result.
Biological response modifiers (BRMs), such as infliximab or adalimumab, may be effective. Other alternatives are golimumab, certolizumab, tocilizumab and rituximab.
Cyclophosphamide should be the first choice in treating patients with associated potentially lethal vasculitic diseases, such as granulomatosis with polyangiitis or polyarteritis nodosa.
Diagnostic biopsy can be helpful.
Surgery can be required to address complications such as cataract formation and intractable glaucoma.
Generally those with a lower degree of inflammation respond to NSAID treatment alone, whilst those with more severe inflammation require steroid treatment. Those with nodular disease (particularly those with associated systemic disease) are more likely to respond to immune-modifying therapies, particularly alkylating agents - for example, methotrexate.21
Necrotising anterior scleritis
This is managed with systemic steroids and immunosuppressive therapies and may need surgical intervention if perforation is impending.23
Cyclophosphamide should be the first choice in treating patients with an underlying systemic vasculitis such as granulomatosis with polyangiitis or polyarteritis nodosa.
In case of therapeutic failure, BRMs such as infliximab or adalimumab may be effective. Other alternatives are golimumab, certolizumab, tocilizumab and rituximab.24
Periocular steroid injections should not be used in necrotising scleritis; they may exacerbate the condition.
Pulsed intravenous cyclophosphamide with or without pulsed intravenous corticosteroids may be required for urgent cases.
Scleromalacia perforans25
There is no specific effective treatment.
Topical treatment is insufficient.
Topical ciclosporin A has been tried, as have immunosuppressants, biological agents and tumour necrosis factor inhibitors.
Surgical treatment may be needed to preserve globe integrity where the uvea is exposed. Patch grafts or pedicle flaps may be useful.
Posterior scleritis
Younger patients usually respond well to oral NSAIDs but elderly patients with associated disease or patients in whom there is a threat to vision are managed as with anterior non-necrotising scleritis.26
There are emerging treatment modalities such as infliximab.27 However, these remain the remit of sub-specialist and research centres.
Scleritis - complications
Back to contentsScleral thinning.
Ischaemia of the anterior segment of the globe.
Raised intraocular pressure.
Retinal detachment.
Uveitis.
Cataract.
Phthisis (globe atrophy).
Scleritis - prognosis
Back to contentsWhen scleritis originates from a systemic disorder, its course tends to follow that of the underlying problem.
Where there is non-necrotising scleritis, the visual prognosis is usually reasonable providing that there are no ocular complications (for example, uveitis).
Anterior necrotising scleritis with inflammation is the most severe and distressing form of the disease. Most patients have an associated vascular disease and this also carries a significant mortality. Visual prognosis is poor.
Posterior scleritis, in patients aged over 50, is associated with an increased risk of harbouring systemic disease and suffering visual loss.
Scleritis - prevention
Back to contentsThere are no clear-cut preventative measures other than the rigorous control of the progression of any underlying disease and early treatment of the scleritis to minimise the risk of visual loss.
Dr Mary Lowth is an author or the original author of this leaflet.
Further reading and references
- Dutta Majumder P, Agrawal R, McCluskey P, et al; Current Approach for the Diagnosis and Management of Noninfective Scleritis. Asia Pac J Ophthalmol (Phila). 2020 Dec 7;10(2):212-223. doi: 10.1097/APO.0000000000000341.
- Murthy SI, Sabhapandit S, Balamurugan S, et al; Scleritis: Differentiating infectious from non-infectious entities. Indian J Ophthalmol. 2020 Sep;68(9):1818-1828. doi: 10.4103/ijo.IJO_2032_20.
- Moshirfar M, Ronquillo Y; Infectious Scleritis.
- Jabs DA, Mudun A, Dunn JP, et al; Episcleritis and scleritis: clinical features and treatment results. Am J Ophthalmol. 2000 Oct;130(4):469-76.
- Berchicci L, Miserocchi E, Di Nicola M, et al; Clinical features of patients with episcleritis and scleritis in an Italian tertiary care referral center. Eur J Ophthalmol. 2014 May-Jun;24(3):293-8. doi: 10.5301/ejo.5000394. Epub 2013 Nov 12.
- Watson PG, Hayreh SS; Scleritis and episcleritis. Br J Ophthalmol. 1976 Mar;60(3):163-91.
- Williamson J; A Red Eye: Scleritis or Episcleritis?, Review of Optometry, 2017.
- Sen HN, Sangave AA, Goldstein DA, et al; A standardized grading system for scleritis. Ophthalmology. 2011 Apr;118(4):768-71. Epub 2010 Nov 20.
- McCluskey P, Wakefield D; Prediction of response to treatment in patients with scleritis using a standardised scoring system. Aust N Z J Ophthalmol. 1991 Aug;19(3):211-5.
- Honik G, Wong IG, Gritz DC; Incidence and prevalence of episcleritis and scleritis in Northern California. Cornea. 2013 Dec;32(12):1562-6. doi: 10.1097/ICO.0b013e3182a407c3.
- Saikia P, Nashed A, Helbig H, et al; Bilateral posterior scleritis: an idiopathic painless presentation. Ocul Immunol Inflamm. 2010 Dec;18(6):452-3. Epub 2010 Oct 10.
- Elnahry AG, Elnahry GA; Granulomatous Uveitis.
- Schonberg S, Stokkermans TJ; Episcleritis
- Shields MK, Furtado JM, Lake SR, et al; Syphilitic scleritis and episcleritis: A review. Asia Pac J Ophthalmol (Phila). 2024 May-Jun;13(3):100073. doi: 10.1016/j.apjo.2024.100073. Epub 2024 May 23.
- Gungor K, Bekir NA, Namiduru M; Recurrent episcleritis associated with brucellosis. Acta Ophthalmol Scand. 2001 Feb;79(1):76-8.
- Akpek EK, Thorne JE, Qazi FA, et al; Evaluation of patients with scleritis for systemic disease. Ophthalmology. 2004 Mar;111(3):501-6.
- Lagina A, Ramphul K; Scleritis
- Gordon, L. Orbital inflammatory disease: a diagnostic and therapeutic challenge. Eye 20, 1196–1206. 2006.
- Rich RM, Smiddy WE, Davis JL; Infectious scleritis after retinal surgery. Am J Ophthalmol. 2008 Apr;145(4):695-9. Epub 2008 Feb 1.
- Sawant SD, Biswas J; Fungal scleritis with exudative retinal detachment. Ocul Immunol Inflamm. 2010 Dec;18(6):457-8. Epub 2010 Sep 16.
- Thakker MM, Perez VL, Moulin A, et al; Multifocal nodular episcleritis and scleritis with undiagnosed Hodgkin's lymphoma. Ophthalmology. 2003 May;110(5):1057-60.
- Ramirez-Ortiz MA, Vasquez-Resendis A; Isolated bilateral posterior scleritis after eye trauma. J AAPOS. 2007 Jun;11(3):284-5. Epub 2007 Jan 25.
- Etminan M, Forooghian F, Maberley D: CMAJ. 2012 May 15; 184(8): E431–E434. Inflammatory ocular adverse events with the use of oral bisphosphonates: a retrospective cohort study
- Sainz de la Maza M, Molina N, Gonzalez-Gonzalez LA, et al; Scleritis therapy. Ophthalmology. 2012 Jan;119(1):51-8. doi: 10.1016/j.ophtha.2011.07.043. Epub 2011 Oct 19.
- Abdel-Aty A, Gupta A, Del Priore L, et al; Management of noninfectious scleritis. Ther Adv Ophthalmol. 2022 Jan 21;14:25158414211070879. doi: 10.1177/25158414211070879. eCollection 2022 Jan-Dec.
- Aklek E; Necrotizing scleritis; Diagnosis and Therapy, Uveitis.org, 2017.
- Onal S, Kazokoglu H, Koc A, et al; Rituximab for remission induction in a patient with relapsing necrotizing scleritis associated with limited Wegener's granulomatosis. Ocul Immunol Inflamm. 2008 Sep-Oct;16(5):230-2.
- Kopacz et al.; Scleromalacia Perforans: What We Know and What We Can Do. J Clinic Experiment Ophthalmol 2013, S2
- Beardsley RM, Suhler EB, Rosenbaum JT, et al; Pharmacotherapy of scleritis: current paradigms and future directions. Expert Opin Pharmacother. 2013 Mar;14(4):411-24. doi: 10.1517/14656566.2013.772982. Epub 2013 Feb 21.
- Sassa Y, Kawano YI, Yamana T, et al; A change in treatment from etanercept to infliximab was effective to control scleritis in a patient with rheumatoid arthritis. Acta Ophthalmol. 2011 Apr 6. doi: 10.1111/j.1755-3768.2010.02090.x.
Continue reading below
Article history
The information on this page is written and peer reviewed by qualified clinicians.
Next review due: 16 Jun 2028
17 Jun 2025 | Latest version

Ask, share, connect.
Browse discussions, ask questions, and share experiences across hundreds of health topics.

Feeling unwell?
Assess your symptoms online for free