Thalassaemia
Peer reviewed by Dr Doug McKechnie, MRCGPLast updated by Dr Colin Tidy, MRCGPLast updated 23 Feb 2023
Meets Patient’s editorial guidelines
- DownloadDownload
- Share
- Language
- Discussion
- Audio Version
- Add to preferred sources on Google
Thalassaemia is an inherited condition affecting the blood. There are different types, which vary from a mild condition to a serious or life-threatening condition. For the more severe forms of thalassaemia, modern treatment gives a good outlook, but lifelong monitoring and treatment are needed. Good treatment is important to prevent complications developing.
At a glance
Thalassaemia is an inherited blood condition affecting haemoglobin, the chemical that carries oxygen in the blood.
It is caused by genetic changes that mean part of the haemoglobin is faulty, leading to anaemia.
There are different types of thalassaemia, ranging from mild with no symptoms to severe, lifelong conditions requiring treatment.
Thalassaemia major is a severe type, often needing regular blood transfusions and chelation treatment to manage iron overload.
Thalassaemia is more common in people whose family origins are Mediterranean, Middle Eastern, or Asian.
A blood test can diagnose thalassaemia, and screening is offered to pregnant women and newborn babies in the UK.
What is thalassaemia?
Thalassaemia is an inherited (genetic) condition affecting the blood. There are different types of thalassaemia. Depending on which type you have, thalassaemia may cause no illness at all, or may be a serious lifelong condition requiring treatment.
What causes thalassaemia?
The cause is an inherited (genetic) change, involving the genes which tell the body how to make an important chemical called haemoglobin. Haemoglobin is the chemical which carries oxygen in the blood - it is the one which gives blood its red colour. Haemoglobin is located in cells called red blood cells which are part of the blood.
Haemoglobin is made out of different parts. The main parts are called alpha chains and beta chains which are put together to make the haemoglobin molecule. In thalassaemia, part of the haemoglobin is faulty - usually either the alpha chains or the beta chains. This means that some of the haemoglobin does not work properly.
As a result, there is not enough normal haemoglobin and the red blood cells break down easily. This makes the person lacking in haemoglobin (anaemic), with various symptoms. Meanwhile, the body tries to make more haemoglobin and more red blood cells. So, the blood system goes into overproduction mode which can cause more symptoms and complications.
Depending on the type of thalassaemia, the amount of abnormal haemoglobin varies. It can be most of the body's haemoglobin, or only a small proportion. This is mainly what determines how severe the thalassaemia is. There are also other individual factors involved. So, two people with the same type of thalassaemia may have a different severity of illness from the same condition.
What are the different types of thalassaemia?
The main types of thalassaemia are called alpha thalassaemia and beta thalassaemia. (The alpha and beta refer to which haemoglobin gene is affected, and which of the haemoglobin chains is faulty.) There are some rarer types too.
Each type of thalassaemia (alpha and beta) is then classified into more types, according to how severe the condition is. This mainly depends on how many thalassaemia genes are involved.
The mildest types are called thalassaemia trait (or thalassaemia minor).
The more severe beta types are beta thalassaemia major (BTM) and beta thalassaemia intermedia (BTI).
The more severe alpha forms are Hb Barts (very severe) and HbH disease (moderate).
There are also some rarer types of thalassaemia such as delta beta thalassaemia, or combinations of a beta-thalassaemia gene with another abnormal haemoglobin gene such as HbE.
Thalassaemia trait
This means that you carry a thalassaemia gene but can still make enough normal haemoglobin. So, you will usually not have any or just relatively mild symptoms or problems from the thalassaemia. You may not know you have it unless you have a special blood test. However, it can be useful to know your diagnosis because:
Some types of thalassaemia trait give you a very mild type of anaemia, where your red blood cells are smaller and paler than usual (described in laboratory reports as 'microcytic and hypochromic'). This can be mistaken for iron deficiency.
Your children can inherit the gene. By itself this is not a problem. However, if your partner also has a similar gene, your children might get a double dose of the abnormal haemoglobin gene and could inherit a severe form of thalassaemia. It is possible to arrange tests for parents or for an unborn baby, to see whether the baby could be affected.
The different types of thalassaemia trait are:
Alpha plus thalassaemia trait:
Alpha plus thalassaemia trait. This means that you have one missing alpha haemoglobin gene. (Normally there are four of these genes.) This trait can ONLY cause a problem if your partner has alpha zero thalassaemia trait - in which case your children might inherit HbH disease (explained below). Apart from that situation, it will not affect you or your children.
Alpha zero thalassaemia trait. This means you have two missing alpha haemoglobin genes (out of the normal four alpha genes). It will not make you ill, but if your partner also has alpha zero thalassaemia trait, your children might inherit a severe condition called Hb Barts (explained below). Or, if your partner has alpha plus thalassaemia trait then your children might inherit HbH disease (see below).
Beta-thalassaemia trait. This means you have one abnormal beta-haemoglobin gene (out of the normal two beta genes). It will not make you ill. But, if your partner also has beta-thalassaemia trait then your children could inherit BTM or BTI (see below). Beta-thalassaemia trait can also interact with other abnormal haemoglobin genes which are not thalassaemias. For example, if your partner has a gene for sickle cell anaemia then your children might inherit a serious condition called sickle cell/beta thalassaemia (see below).
Thalassaemia major
A person with beta thalassaemia major (BTM) has two beta-thalassaemia genes (ie two abnormal beta-haemoglobin genes). Most of their haemoglobin is abnormal and does not work. This causes severe anaemia starting around the age of 4-6 months. Before that, the baby is not affected. This is because until age 3-6 months the baby makes a different type of haemoglobin, called fetal haemoglobin, which is not affected by the thalassaemia gene. With BTM, you need regular blood transfusions, plus other treatment to prevent complications.
Beta thalassaemia intermedia (BTI)
As the name suggests, this type is less severe than BTM. You have two beta-thalassaemia genes but can make some haemoglobin which works reasonably well. This may be because your particular combination of thalassaemia genes is (in effect) less severe, or because of some other protective factor. Although less severe than thalassaemia major, thalassaemia intermedia does need regular monitoring for life and often needs some treatment to prevent complications.
Sickle cell/beta thalassaemia
This can occur if one parent has a beta-thalassaemia gene, and the other parent carries a gene for a different haemoglobin disorder called sickle cell anaemia. If their child inherits one of each gene, the combination is called sickle cell/beta thalassaemia - also called sickle cell disease. This condition behaves like sickle cell anaemia (not like thalassaemia) and is treated in the same way as sickle cell anaemia. See the separate leaflet called Sickle Cell Disease (Sickle Cell Anaemia) for more detail.
HbH disease
This is a type of alpha thalassaemia. It is due to having three missing alpha-haemoglobin genes (normally each person has four of these genes). This can happen if one parent has alpha plus thalassaemia and the other has alpha zero thalassaemia. It usually causes a mild but persistent anaemia. Sometimes HbH causes more symptoms and is similar to BTI (explained below). Some people with HbH disease need blood transfusions.
Hb Barts
This is the most severe form of thalassaemia, where all the alpha-haemoglobin genes are abnormal or missing. It occurs if a baby inherits two alpha zero thalassaemia genes. In this condition, no normal haemoglobin can be made, even before birth. It is the most serious form of thalassaemia - so serious that the baby will usually die in the womb from severe anaemia. There have been rare cases where the baby has been saved by blood transfusions being given in the womb, with the transfusions then continuing after birth.
How common is thalassaemia?
Worldwide, thalassaemia is one of the most common inherited diseases. However the number of people with thalassaemia is very variable between people from different parts of the world. It is much more common in people from the Mediterranean, Middle East, Central Asia, Indian Subcontinent, Far East and Africa. The highest recorded rates occur in Cyprus and Sardinia.
Beta thalassaemia is common in areas around the Mediterranean, in the Middle East, in Central, South and Southeast Asia, and in Southern China.
Alpha thalassaemia is common in Southeast Asia, Africa, and India.
The World Health Organization (WHO) estimates that about 3 people in every 200 of the world's population may be beta thalassaemia carriers and that at least 60,000 severely affected people are born each year.
How is thalassaemia inherited?
A child inherits haemoglobin genes from both parents. For example, if both parents have beta-thalassaemia trait, there is: a 1 in 4 chance of the child having normal haemoglobin genes; a 1 in 2 chance of the child having beta-thalassaemia trait; and a 1 in 4 chance the child will have BTM or BTI.
Diagram outlining thalassaemia inheritance
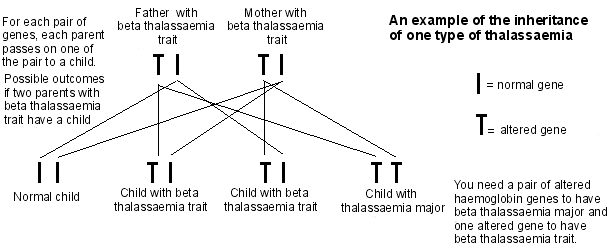
Who gets thalassaemia?
Anyone may carry a thalassaemia gene. On average, 3 in 100 of the world's population have a thalassaemia gene (and therefore have a thalassaemia trait). The chance of having a thalassaemia gene varies, depending on your family origin.
Thalassaemia is most common in people whose family origins are Mediterranean or Asian. It can be diagnosed from a blood test. For example, beta thalassaemia genes are carried by: 1 in 7 Greek Cypriots, 1 in 12 Turks, 1 in 20 Asians, 1 in 20-50 Africans/Afro-Caribbeans (depending on which part of Africa your family comes from) and 1 in 1,000 English of North European origin.
Pregnant women and couples planning a family are advised to have a test for thalassaemia, because early diagnosis can be helpful. In England, all pregnant women and newborn babies are now offered a thalassaemia test, but you can ask for a test before you become pregnant.
Thalassaemia test
The diagnosis is made by a blood test. The blood sample is analysed to see what type of haemoglobin is present in the blood.
In some cases, extra tests such as DNA (genetic) tests are needed to diagnose the exact type of thalassaemia. It may help to test other family members where possible.
Thalassaemia trait may be suspected from the results of an ordinary blood test called a full blood count. If the result shows red blood cells that are smaller and paler than usual, this may be due to iron deficiency or to thalassaemia trait.
Tests for pregnant women
Policies for screening pregnant women and babies vary throughout the UK - see the UK Screening Portal link under 'Further Reading and References', below. If the result shows that you carry a thalassaemia gene then a test is also offered to the baby's father (if possible). The results of both parents' tests will help to decide whether the baby could be affected by thalassaemia. The results will be explained to you.
If there is a chance that the baby could be affected, you will be offered counselling to discuss whether you would like to have a further test for the unborn baby (a prenatal test). This test finds out whether the unborn baby actually has thalassaemia - and which type. You will be given information about how this could affect the baby. If the baby has a severe form of thalassaemia, you will be offered counselling to discuss whether you want to continue with the pregnancy.
When is the best time to have a test?
If you are pregnant, the best time to have a thalassaemia blood test for yourself is before you are 10 weeks pregnant. This allows more time to test your partner or your baby, if needed. You can ask your doctor for a test early in pregnancy, if it is not already offered to you at that time. However, tests can still be done at a later stage.
A prenatal test (on the unborn baby) can be done from 10 weeks of pregnancy onwards, depending on the type of test used. The usual tests offered are chorionic villus sampling (CVS) or amniocentesis.
Should I and my partner have a thalassaemia test before starting a family?
Women or couples may want to have tests for thalassaemia before starting a family, especially if their family origins make thalassaemia more likely. The UK Thalassaemia Society and many health professionals encourage awareness of thalassaemia and early testing. The test can be arranged by your doctor.
The advantage of having tests before you become pregnant, is that you will know whether or not there is a possibility that your baby could inherit a severe form of thalassaemia. This may be helpful when making decisions about pregnancy. For example, you may want to have a prenatal test during pregnancy if there is a risk of a severe condition for the baby.
Tests for newborn babies
In the UK, all newborn babies are offered a bloodspot test at 5-8 days after birth. This tests for a number of medical conditions, which are considered important because early treatment makes a difference. The test is done by taking a small spot of blood from the baby's heel.
Throughout the UK, the bloodspot test now includes testing for thalassaemia and other haemoglobin disorders such as sickle cell disease. Policies for screening newborn babies vary throughout the UK - see the UK Screening Portal link under 'Further Reading and References', below.
The bloodspot test will diagnose most types of thalassaemia. You will be given the results about six weeks later. If the baby has thalassaemia trait, no action or treatment is needed. If the baby has a more severe type of thalassaemia which needs treatment, the result will be explained. You will be given a clinic appointment so that the diagnosis can be checked, and treatment can be started if necessary.
More about beta thalassaemia
The rest of this leaflet explains more about beta thalassaemia major (BTM) and beta thalassaemia intermedia (BTI). These are the more common types of thalassaemia needing treatment.
Beta thalassaemia major
Thalassaemia symptoms
Ideally, BTM will be diagnosed and treated early in order to prevent symptoms and reduce complications. So with good care, there may be few symptoms.
If untreated, symptoms of BTM start at around age 4-6 months. Symptoms come on gradually and are:
Thalassaemia anaemia - the baby may be pale, lacking in energy (lethargic), not feeding or growing well, and prone to infections.
Bone symptoms - the body tries to produce more red blood cells. This is a natural reaction to anaemia. However, it does not help thalassaemia much because most of the haemoglobin that is produced is abnormal. The result is over-expansion of the bone marrow, which is the body's blood cell factory. This affects bone growth including the face and jaw bones, making the forehead and upper jaw very prominent.
Without treatment, symptoms of BTM become gradually worse. Untreated, children with BTM usually die from infection or heart failure in childhood.
Can thalassaemia be cured?
A possible cure is having a transplant of stem cells. This means either a bone marrow transplant, or a cord blood transplant. These treatments take normal blood-making cells from a donor, and give them to the person with thalassaemia. If the transplant is successful, these cells last for life and make normal haemoglobin - a lifelong cure.
However, a stem cell transplant is not suitable for everyone. You need a suitable donor, and there are some serious risks involved. UK guidelines recommend that all BTM patients have the opportunity to discuss stem cell transplantation with a specialist.
How is BTM treated?
There are two main treatments: blood transfusions and chelation treatment.
Blood transfusions are started if you (or your child) have anaemia plus other symptoms, such as poor growth, not feeding well or other problems. If you only have anaemia and are otherwise doing well, your doctor may advise just monitoring the situation for a time. This is because some people thought to have BTM turn out to have the milder condition of BTI, and may not need transfusions.
Blood transfusions give normal red blood cells to the person with BTM. This corrects the anaemia for a while, which improves health and helps children to grow normally. However, the red blood cells have a limited lifespan. So, transfusions normally have to be repeated every 3-4 weeks.
Chelation treatment is important, to remove iron from the body. With thalassaemia, the body gets overloaded with iron. This is partly from blood transfusions, and also because the thalassaemia itself makes the body take up (absorb) more iron from food. If the excess iron is not removed, it can damage internal organs and cause complications. Chelation helps the body get rid of excess iron. This treatment is really important for preventing complications.
There are different forms of chelation: deferiprone (Ferriprox®) and deferasirox (Exjade®), which are taken by mouth; however, desferrioxamine (Desferal®) is given via a drip (an infusion inserted under the skin). Each form of chelation has its pros and cons, and sometimes combinations are used. Your doctor can discuss the options and help to decide which is most suited to you. Chelation is usually started within a year or two after starting transfusions.
Treatment for BTM should be given by a specialist team who have experience in treating thalassaemia. If this is difficult because of where you live then a specialist should advise the doctors who are treating you, and you should be seen by a specialist at least once a year. Regular reviews and check-ups are really important, to ensure that your treatment matches your needs, to check for side-effects and to prevent complications.
A healthy lifestyle is also advised for thalassaemia. Avoid smoking and excess alcohol. Good nutrition and regular exercise can help. You may need extra vitamins, such as folic acid, vitamin D and zinc.
What are the possible complications of BTM and how are they prevented or treated?
Complications of anaemia and transfusions
Untreated anaemia can affect growth and bone development because the bone marrow expands to try to make more blood cells. Anaemia can also cause an enlarged spleen (the spleen is an organ in the tummy (abdomen) which is part of the immune system). A large spleen can make anaemia worse, so you may need an operation to remove the spleen. If your spleen is removed, you will need extra immunisations and daily penicillin, to protect against certain infections (pneumococcal infection and meningitis).
Transfusions can cause blood reactions. These are less likely if the blood is very carefully matched to be as close as possible to your blood type. Infections such as hepatitis B and hepatitis C can be transmitted by transfusion. This is less likely in the UK and countries where donor blood is tested for infections. Hepatitis B immunisation is also recommended.
Complications of iron overload
Before chelation treatment, iron overload was a major problem for people with BTM. Chelation has reduced the complications of iron overload but they can still occur. So, you will need regular monitoring to check iron levels and possible complications.
Iron overload can damage various organs in the body - for example, the heart, liver, hormone glands, pancreas (causing diabetes) and bones. So you will need regular blood tests and scans to check the function of these organs. For children, growth and development are also monitored. If some organs are affected by iron overload, you may need increased chelation treatment or other treatments. If the hormone glands are affected, replacement hormones can be taken.
Complications of chelation
Chelation treatments have various possible side-effects. The side-effects may involve the blood, liver, kidneys, vision, hearing and bones. So, if you are having chelation, you will need regular blood and urine tests, plus checks for eyes, hearing and a child's growth. If you develop side-effects with chelation, the dose may need adjusting, or you may need a different chelator.
Infections
People with BTM can be more prone to serious bacterial infections (for various reasons). So, obtain medical advice quickly if you feel more unwell than usual, or if you have symptoms of infection such as a high temperature (fever). Certain types of infection (from species of germs (bacteria) called Yersinia and Klebsiella) are more common than usual, due to iron overload or chelation treatment. Yersinia causes tummy pain, diarrhoea and fever. Sometimes, this can mimic appendicitis. Klebsiella causes fever and severe illness. So obtain medical advice urgently if you have these symptoms. Tell doctors and nurses about your thalassaemia and the treatment you are taking. Non-specialist doctors should also contact your thalassaemia specialist for advice if you are unwell.
Bone problems
With BTM, bone problems can occur, due to the thalassaemia itself or from chelation treatment. Also, 'thinning' of the bones (osteoporosis) can occur at a younger age than usual. So, your growth (if a child) and bone health will need checking. A good intake of vitamin D and calcium helps to prevent osteoporosis. Various medications can help to treat osteoporosis. If your bones are severely affected, you may need specialist advice.
What is the outlook for BTM?
Untreated, BTM is a severe illness with worsening anaemia, infections and heart failure. Without treatment, this usually leads to death by the age of 5 years. With treatment, the outlook (prognosis) is good because anaemia and complications can be controlled by transfusions and chelation treatment. Nowadays, treatment of thalassaemia is usually successful, with patients living into adulthood and generally able to have careers, relationships and children.
The long-term outlook depends on how well complications can be prevented, particularly the iron overload. Early deaths can still occur, and children sometimes develop complications such as poor growth. What makes the most difference to the outlook is good chelation treatment. Also, patients treated at specialist centres have had better outcomes. Specialist treatment is now recommended for all thalassaemia patients in the UK.
The outlook will probably continue to improve, because of recent progress in chelation treatment.
About beta thalassaemia intermedia
BTI is like a milder form of BTM. The body can produce some functioning haemoglobin, but not as much as normal. So, you have a moderate level of anaemia, but the body can usually adjust to it without needing regular transfusions. You need to take an extra vitamin called folic acid, which helps the body make blood cells.
As with BTM, you will need regular check-ups and specialist advice, to monitor the anaemia and prevent complications.
Blood transfusions may be needed sometimes. For example, during pregnancy, before surgery, or if you have an infection (some infections can make the anaemia worse); also, if there are symptoms of anaemia, such as poor growth. Some people develop leg ulcers (due to anaemia), which can also be helped by transfusion.
Because of the anaemia, your spleen may enlarge, and an operation to remove the spleen may help (as explained above for BTM). Another possible treatment for the anaemia is a medication called hydroxycarbamide (hydroxyurea). It may help some patients but the benefits are uncertain and it can have serious side-effects.
People with BTI can have iron overload, similar to patients with BTM, although it may be less severe. This can happen, even if you do not have transfusions, because the thalassaemia causes extra iron to be absorbed from food. As with BTM, chelation treatment is important to remove excess iron and to prevent complications.
Other possible complications of BTI are:
Bone and growth problems (similar to BTM, because the bone marrow over-expands).
Swellings near the spine (which don't usually cause problems, but sometimes press on a nerve and need treatment).
Gallstones.
Your blood may be more prone to forming clots.
Sometimes, a lung complication called pulmonary hypertension.
Patient picks for Blood disorders

Allergies, blood and immune system
Thrombophilia
The body has a natural clotting process in the blood, which is altered in thrombophilia. The normal clotting process is called haemostasis.
by Dr Toni Hazell, FRCGP

Allergies, blood and immune system
Haemophilia
Haemophilia is a rare bleeding disorder that affects your ability to make a clot. The blood contains substances called clotting factors that work with platelets to form a clot. When you injure yourself the clot stops you bleeding. Haemophilia is a condition where there is a deficiency of clotting factors, so people with this condition usually have prolonged bleeding. There is no cure but with available treatments most people with haemophilia are able to live normal lives.
by Dr Hayley Willacy, FRCGP
Frequently asked questions
If I have thalassaemia trait, should my partner be tested?
Yes, if you have been diagnosed with thalassaemia trait, it is recommended that your partner also gets tested. This is important to determine if there's a risk of your children inheriting a severe form of thalassaemia. If both parents carry similar thalassaemia genes, there is a chance their children could receive a double dose of the abnormal gene, leading to a serious condition like beta thalassaemia major or Hb Barts. Knowing this information can help you make informed decisions about family planning and prenatal testing.
What is the best time for a pregnant woman to have a thalassaemia test?
If you are pregnant, the best time to have a thalassaemia blood test for yourself is before you are 10 weeks pregnant. This timing allows for more time if additional tests are needed for your partner or the baby. While early testing is ideal, tests can still be conducted later in the pregnancy.
Can beta thalassaemia intermedia (BTI) get worse and require regular blood transfusions?
BTI is generally a milder form of thalassaemia, where the body can produce some functioning haemoglobin, and regular transfusions are not typically needed. However, blood transfusions may be required occasionally, such as during pregnancy, before surgery, or if you develop an infection that worsens anaemia. They might also be needed if symptoms of anaemia, like poor growth, become significant, or to treat leg ulcers caused by anaemia.
What kind of lifestyle adjustments are recommended for someone with thalassaemia?
For individuals with thalassaemia, a healthy lifestyle is advised. This includes avoiding smoking and excess alcohol. Good nutrition and regular exercise can also be beneficial. You may also need to take extra vitamins, such as folic acid, vitamin D, and zinc, to support your health.
Further reading and references
- NHS Sickle Cell and Thalassaemia Screening Programme; GOV.UK.
- Management of Beta Thalassaemia in Pregnancy; Royal College of Obstetricians and Gynaecologists (Mar 2014)
- Rund D; Thalassemia 2016: Modern medicine battles an ancient disease. Am J Hematol. 2016 Jan;91(1):15-21. doi: 10.1002/ajh.24231.
- Ali S, Mumtaz S, Shakir HA, et al; Current status of beta-thalassemia and its treatment strategies. Mol Genet Genomic Med. 2021 Dec;9(12):e1788. doi: 10.1002/mgg3.1788. Epub 2021 Nov 5.
- Yousuf R, Akter S, Wasek SM, et al; Thalassemia: A Review of the Challenges to the Families and Caregivers. Cureus. 2022 Dec 13;14(12):e32491. doi: 10.7759/cureus.32491. eCollection 2022 Dec.
- Origa R; beta-Thalassemia. Genet Med. 2017 Jun;19(6):609-619. doi: 10.1038/gim.2016.173. Epub 2016 Nov 3.
- Harteveld CL, Higgs DR; Alpha-thalassaemia. Orphanet J Rare Dis. 2010 May 28;5:13. doi: 10.1186/1750-1172-5-13.
About the authorView full bio

Dr Colin Tidy, MRCGP
General Practitioner, Medical Author
MBBS, MRCGP, MRCP (Paediatrics), DCH
Dr Colin Tidy is an NHS Doctor, based in Oxfordshire.
About the reviewerView full bio

Dr Doug McKechnie, MRCGP
Medical Writer
MA, MBBS, MSc, DRCOG, MRCP(UK), MRCGP(2021), FHEA
Dr Doug McKechnie is an NHS GP working in London. He works full-time clinically and is also the Deputy Lead for the Clinical and Professional Practice module at University College London Medical School.
Article history
The information on this page is written and peer reviewed by qualified clinicians.
Article also available in English, German, Spanish, French, Italian, Portuguese, Hindi, Hebrew, Arabic, and Swedish.
Next review due: 22 Feb 2028
23 Feb 2023 | Latest version

Ask, share, connect.
Browse discussions, ask questions, and share experiences across hundreds of health topics.

Feeling unwell?
Assess your symptoms online for free
Sign up to the Patient newsletter
Your weekly dose of clear, trustworthy health advice - written to help you feel informed, confident and in control.
By subscribing you accept our Privacy Policy. You can unsubscribe at any time. We never sell your data.
More in allergies, blood and immune system
- Allergic conjunctivitis
- Antibody and antigen tests
- Antihistamines
- Blood clotting tests
- Bone marrow biopsy and aspiration
- Cardiac enzymes
- Chronic lymphocytic leukaemia
- Chronic myeloid leukaemia
- Deep vein thrombosis
- Diets suitable for people with anaemia
- Folic acid deficiency
- Granulomatosis with polyangiitis
- Hives
- Lupus
- Polycythaemia rubra vera
- Porphyria
- PSA test
- Psoriasis
- Swollen lymph glands
- Thrombophilia