Cystic fibrosis
Peer reviewed by Dr Hayley Willacy, FRCGP Last updated by Dr Toni Hazell, FRCGPLast updated 16 May 2023
Meets Patient’s editorial guidelines
- DownloadDownload
- Share
- Language
- Discussion
- Audio Version
- Add to preferred sources on Google
In this series:Sweat test
Cystic fibrosis is an inherited disease which mainly affects the lungs and pancreas but can involve other organs. Symptoms usually begin in early childhood and include persistent cough, wheeze, repeated chest infections, difficulty digesting food and general ill health.
Treatments include antibiotics, physiotherapy, medicines for thinning mucus, pancreatic enzyme replacements and other therapies.
At a glance
Cystic fibrosis is a genetic condition affecting mainly the lungs and pancreas.
It causes cells to make thicker than normal mucus and secretions.
Symptoms often appear in the first year of life, but severity varies.
Common symptoms include persistent cough, wheezing, and recurring chest infections.
Digestive issues can lead to poor growth and large, smelly stools.
There is no cure, but treatments help manage symptoms and improve outlook.
What is cystic fibrosis?
Cystic fibrosis is a condition which mainly affects the lungs and pancreas but can affect other parts of the body, including the liver, nose and sinuses and sweat glands. Normally, cells in these parts of the body make mucus and other watery juices and secretions.
In people with cystic fibrosis, these cells do not function correctly and make mucus and secretions which are thicker than normal. This can cause various symptoms and problems (which are described below).
Cystic fibrosis symptoms
Symptoms of cystic fibrosis usually first develop within the first year of life but may not appear until later childhood. The severity of symptoms can vary.
Lung symptoms
The cells that line the airways of the lungs make sputum (phlegm) that is thicker than normal and which is not cleared from the lungs easily. This can trap germs (bacteria) in the small airways and lead to infection and inflammation. So, symptoms which typically develop include:
Persistent cough which typically produces a lot of sputum.
Shortness of breath and breathing difficulties.
Recurring chest infections. These can be severe, such as pneumonia. Repeated infections and inflammation can damage the lungs and lead to poor lung function.
Gut symptoms (digestive system)
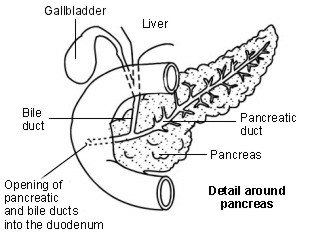
The pancreas normally makes digestive juices which contain chemicals (enzymes). The digestive juices normally flow out from the pancreatic duct into the duodenum and digest food.
Diagram showing detail around the pancreas
Liver function
In people with cystic fibrosis, thickened secretions block the normal flow of the digestive juices from the pancreas. This can result in food not being digested or absorbed properly - in particular, fatty foods and fat-soluble vitamins (vitamins A, D, E and K).
This can cause:
Malnutrition leading to poor growth and poor weight gain (even if you have a good appetite and eat a lot, as the problem is with digesting and absorbing the food).
Large, smelly, greasy, fatty stools (faeces) occur in about a third of cases.
Bloated tummy (abdomen).
In about 3 in 10 cases the pancreas functions well and there are no, or minimal, gut symptoms and mainly just lung symptoms.
Symptoms sometimes occur at birth
About 1 in 10 children with cystic fibrosis are diagnosed shortly after birth. This is due to a condition called meconium ileus where in some cases the gut becomes blocked with meconium. Meconium is a thick, dark, sticky substance which is made by the baby's gut before being born. Urgent surgery may be needed to relieve the blockage.
Other symptoms and complications
Other organs may be affected which may cause various other problems in some cases. Also, the pancreas and airways may become severely affected. Therefore, other problems which may also occur in some cases include:
Infertility (especially in males, as the tubes which carry the sperm can become blocked).
Damage to the liver, which may lead to 'scarring' of the liver (cirrhosis). Liver damage occurs in about 1 in 12 cases (if the small ducts in the liver become blocked or damaged).
Diabetes. (Special cells in the pancreas make insulin. If the pancreas becomes severely damaged over time then insulin levels go down and cystic fibrosis-related diabetes may develop.) This is rare in children but is more common in adults who have had cystic fibrosis.
'Thinning' of the bones (osteoporosis) may develop due to poor absorption of certain foods and, in particular, vitamin D which is needed to maintain healthy bones.
The sweat tastes very salty.
Mild cases
Some cases of cystic fibrosis are diagnosed in adults who have relatively mild symptoms. This may be due to some errors of the cystic fibrosis gene not being as faulty as others. The handling of sodium and chloride may only be mildly affected in these cases.
What causes cystic fibrosis?
Cystic fibrosis is a genetic disorder. A genetic disorder is one that can be passed on from your parents through your genes.
If you have cystic fibrosis, one of your genes does not work properly. This is known as a CFTR gene which is on chromosome 7. There are different errors that can occur in this gene and this means that there are different severities of cystic fibrosis that occur.
The CFTR gene helps to control the way the cells handle salt (sodium and chloride ions). The error in the gene results in the cells being unable to handle sodium and chloride properly.
As a result, cells in affected organs have a fault in the way sodium and chloride travel in and out of the cells. Basically, too much sodium travels into the cells. Water follows the sodium, which leaves too little water outside the cells. This causes the mucus or watery secretions outside the cells to be too thick (for example, in the airways of the lungs).
How is cystic fibrosis inherited?
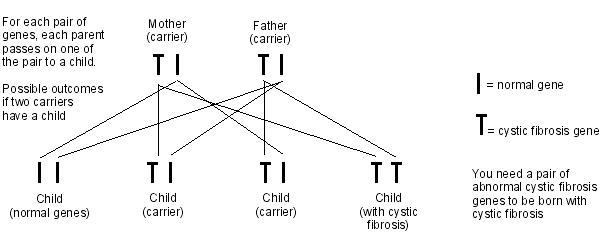
Cystic fibrosis is an autosomal recessive disorder. This means that in order to develop cystic fibrosis you need to inherit two cystic fibrosis genes, one from your mother and one from your father. If you inherit only one cystic fibrosis gene, you are called a carrier.
About 1 in 25 people in the UK of white European descent are carriers of the cystic fibrosis gene. It is much less common in Afro-Caribbean and Asian people. Carriers do not have the disease, as they have one normal gene which can control the salt transport in their cells. But carriers can pass the cystic fibrosis gene on to their children.
When two people who carry the cystic fibrosis gene have a child, there is a:
1 in 4 chance that the child will have cystic fibrosis (by inheriting the cystic fibrosis gene from both parents).
2 in 4 chance that the child will not have cystic fibrosis but will be a carrier (by inheriting a cystic fibrosis gene from one parent but the normal gene from the other parent).
1 in 4 chance that the child will not have cystic fibrosis and will not be a carrier (by inheriting the normal gene from both parents).
Diagram to show cystic fibrosis inheritance
How common is cystic fibrosis?
About 1 in 2,500 babies in the UK are born with cystic fibrosis. Over 9,000 people currently have cystic fibrosis in the UK.
Cystic fibrosis diagnosis
Sweat test
A doctor may arrange a sweat test if he or she suspects cystic fibrosis from the symptoms. This test measures the amount of salt (sodium and chloride) in skin sweat. People with cystic fibrosis have an abnormally high salt level in sweat.
Genetic test
A genetic test can confirm the diagnosis. Some cells are either scraped from the inside of the cheek or taken from a blood test. These can be tested to detect the cystic fibrosis gene.
Screening test
All newborn babies in the UK are now screened for cystic fibrosis. A small heel prick blood test is taken about the sixth day after birth. This can detect a chemical called immunoreactive trypsinogen which is high in babies with cystic fibrosis. If it is high then a sweat test and genetic test can be done to confirm the diagnosis. Screening is considered important because the earlier the diagnosis is made, the sooner treatment can begin which improves the outlook (prognosis).
Other tests
Other tests may be performed to check for the development of complications. These may include X-rays, ultrasound scans and removal of a small amount of body tissue (a biopsy) to examine under a microscope.
Cystic fibrosis treatment
There are many aspects to the treatment of people with cystic fibrosis. Treatment involves the input, advice and expertise of various professionals. These include child health doctors, specialist nurses, physiotherapists, dieticians, counsellors and psychologists as well as your primary healthcare team.
It is usual to have regular checks and tests to monitor the condition and to keep a check on children's growth, development and well-being.
The following list is a brief overview of the commonly used treatments. However, it is not a full or exhaustive account of all the treatments used. An individual treatment plan is needed for each case to take into account individual circumstances.
TREATMENTS FOR LUNG PROBLEMS
Physiotherapy and exercise
Regular chest physiotherapy is very important. This helps to clear the airways of the thick, sticky mucus. A physiotherapist usually shows parents how to do this for their children. It involves a special way to pat the chest firmly whilst the child is lying head-down to encourage sputum to be coughed out. Twice-daily chest physiotherapy is common practice. This may need to be increased during times of chest infections.
There is a wide variety of airway clearance techniques available and your physiotherapist will be able to advise you on the most suitable technique or techniques for you. These are likely to change though as you become older or as your disease changes with time.
It is also important to encourage children to exercise and to be as active and fit as possible. So, sports and games such as running, swimming, football and tennis are encouraged.
Antibiotics and antifungals
Courses of antibiotics are a mainstay of treatment. Many children with cystic fibrosis take regular long-term antibiotics. The dose is increased and/or other types of antibiotics are given when a chest infection develops. Various germs (bacteria) can cause infections and the antibiotics chosen depend on which bacteria are found in samples of sputum.
Antibiotics given into a vein (intravenously) are often required for severe infections that are not controlled with antibiotic tablets. They can also be given by a nebuliser. A nebuliser allows a medicine to be inhaled as a fine mist, through a mask.
A bacterium called Pseudomonas aeruginosa commonly persists in the thick mucus in the airways. To keep this from flaring up into repeated infections, an antibiotic given by nebuliser or inhaler is a common treatment.
Sometimes the lungs become infected with a fungus and antifungal medication is required.
Inhalers
Inhalers to open up the airways as much as possible may be used - for example, salbutamol. This is similar to the treatment used for asthma.
Dornase alfa
This is a medicine given by nebuliser in some cases. It helps to break down and to thin the thick mucus, making it easier to cough up and clear the mucus from the airways. It may reduce the number of lung infections and help to improve lung function.
Oxygen
People with advanced lung disease may benefit from oxygen, particularly overnight.
Other medication to improve lung function - for example, ibuprofen and azithromycin - may also be recommended in some cases.
TREATMENTS FOR PANCREATIC PROBLEMS
Nutrition
The chemicals (enzymes) needed to digest food are greatly reduced in most people with cystic fibrosis. Therefore children with cystic fibrosis need a high fat and carbohydrate diet. A dietician will usually give detailed advice. High-energy drink supplements may also be needed. In addition, vitamin supplements are needed, as many vitamins in food are not absorbed very well. Being well nourished will also help you to fight any chest infections.
Enzyme supplements
In most cases, enzyme supplements are needed to help to digest food. (These replace the enzymes which normally come from the pancreas.) You need to take these supplements every time you eat food. This can mean taking many doses each day.
OTHER TREATMENTS
A range of other problems which are related to cystic fibrosis may develop in some cases and require treatment. For example:
Salt depletion may occur in hot weather and may require salt supplements.
Liver problems develop in some cases and may require specialist liver treatments.
If diabetes develops, it usually requires insulin or tablet treatment.
Nasal growths (polyps) sometimes develop and can be treated with steroid nasal drops and sprays.
Acid reflux from the stomach into the gullet (oesophagus) is common and can be treated with medicines which reduce the acid content of the stomach juices.
Constipation is quite common and may require regular laxatives.
All people with cystic fibrosis should be up to date with routine immunisations and have an annual flu jab to prevent influenza. It is also important to be immunised with pneumococcal vaccine to help prevent pneumonia caused by this bacterium and people with cystic fibrosis were eligible for the COVID-19 vaccination, but are not included in the 2023 Spring booster campaign. It remains to be seen if ongoing COVID-19 vaccination will be offered to this group.
Some children are tested for their immunity against chickenpox (varicella). If you do not have immunity to it then you may be offered the vaccine against chickenpox.
Lung or heart/lung transplantation may be offered in some cases as the lung disease becomes more severe.
Newer treatments are being researched and developed and, if found successful, may become more widely used in the future. For example:
Gene therapy. This involves using an inhaled spray to deliver normal copies of the cystic fibrosis gene to the lungs.
Medicines are being tested which may correct the abnormal salt and water regulation of cells that leads to thickened mucus and secretions being made in the lungs and other organs.
New methods to improve the action of the current treatments are being developed.
What is the outlook (prognosis)?
Treatments for cystic fibrosis have improved dramatically over the past few decades. However it is a lifelong condition and there is currently no cure for cystic fibrosis.
There will be times when symptoms are more severe - mainly when a chest infection develops. Even with treatment, the main risks are recurring chest infections and pneumonia. This can have a repeated damaging effect on lung function which can get worse over time. Most people with cystic fibrosis die of lung complications, mainly respiratory and heart failure.
Cystic fibrosis life expectancy
With improved treatment there has been a dramatic increase in the survival of people with cystic fibrosis over a period of 20 years or so.
In the 1960s and before, most babies born with cystic fibrosis only survived for a few months or years. Today, many people with cystic fibrosis are living into their late 30s and beyond. With optimal care and treatment, it is estimated that over half of today's children with cystic fibrosis should live into their mid 40s or 50s. With treatment, most people with cystic fibrosis can live reasonably normal and productive lives.
However, death in childhood or early adulthood is still not uncommon.
Genetic counselling for cystic fibrosis
People with a family history of cystic fibrosis may wish to have genetic counselling and testing to find out their risk of passing the condition on to their children. A simple test can be done to look at the genes from cells from the inside of the cheek or from blood. The test can detect the cystic fibrosis gene which can show if you are a carrier of the abnormal gene.
Patient picks for Genetic conditions

Children's health
Congenital heart disease
Congenital heart disease is a condition where an abnormality (defect) develops in the heart before birth. There are a number of types of congenital heart defect. Some are mild and cause few problems; others are life-threatening for the baby.
by Dr Mary Harding, MRCGP

Children's health
Cri du chat syndrome
Cri du chat syndrome is a chromosome problem caused by a missing piece of chromosome 5. The syndrome is called cri du chat (French for cry of the cat) because affected babies often have a high-pitched cry. Not all babies with the missing piece of chromosome 5 will develop cri du chat syndrome. Cri du chat syndrome may cause a variety of abnormalities, especially affecting the head and face. Other features may include learning difficulties and slow growth and development. There is no specific treatment. However, physiotherapy, speech and language therapy, and surgical treatment for some abnormal features may be needed. Many affected children will survive well into adulthood. However, those babies severely affected may die within the first year of life.
by Dr Hayley Willacy, FRCGP
Frequently asked questions
What happens if the pancreas or airways become severely affected by cystic fibrosis?
If the pancreas and airways become severely affected by cystic fibrosis, other problems can develop. These may include repeated sinus infections, growths (polyps) in the nose, infertility (especially in males due to blocked tubes carrying sperm), liver damage leading to scarring (cirrhosis), diabetes, inflammation of the pancreas (pancreatitis), and rectal prolapse. Bone thinning (osteoporosis) can also occur due to poor absorption of vitamin D.
Can cystic fibrosis be diagnosed in adults even if symptoms are mild?
Yes, some cases of cystic fibrosis are diagnosed in adults who have relatively mild symptoms. This can happen if the errors in the cystic fibrosis gene are not as severe as others, leading to only a mild effect on how the body handles sodium and chloride.
How often do individuals with cystic fibrosis need to take enzyme supplements?
Most people with cystic fibrosis need to take enzyme supplements every time they eat food. This can mean taking many doses throughout the day to help digest food, as their pancreas may not produce enough of the necessary digestive chemicals.
Are there any specific vaccines recommended for people with cystic fibrosis?
Yes, it is important for people with cystic fibrosis to be up-to-date with routine immunisations. They should also have an annual flu jab and be immunised with the pneumococcal vaccine to help prevent pneumonia. Additionally, some children are tested for chickenpox immunity, and if they lack it, they may be offered the chickenpox vaccine.
What kind of specialists are involved in treating cystic fibrosis?
The treatment for cystic fibrosis involves a team of various professionals. These include child health doctors, specialist nurses, physiotherapists, dieticians, counsellors, psychologists, and the primary healthcare team. An individual treatment plan is created for each person to address their specific needs.
What new treatments are being explored for cystic fibrosis?
New treatments for cystic fibrosis are being researched and developed. These include gene therapy, which uses an inhaled spray to deliver normal copies of the cystic fibrosis gene to the lungs. Also, medicines are being tested that aim to correct the abnormal salt and water regulation in cells, which causes thick mucus. Additionally, new methods to enhance the effectiveness of existing treatments are being developed.
Further reading and references
- Standards of care; Cystic Fibrosis Trust
- Colistimethate sodium and tobramycin dry powders for inhalation for treating pseudomonas lung infection in cystic fibrosis; NICE Technology appraisal guidance, March 2013
- Conway S, Balfour-Lynn IM, De Rijcke K, et al; European Cystic Fibrosis Society Standards of Care: Framework for the Cystic Fibrosis Centre. J Cyst Fibros. 2014 May;13 Suppl 1:S3-22. doi: 10.1016/j.jcf.2014.03.009.
- Cystic fibrosis: diagnosis and management; NICE Guideline (Oct 2017)
- Langton Hewer SC, Smyth AR; Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis. Cochrane Database Syst Rev. 2017 Apr 25;4:CD004197. doi: 10.1002/14651858.CD004197.pub5.
- Lynch JP 3rd, Sayah DM, Belperio JA, et al; Lung transplantation for cystic fibrosis: results, indications, complications, and controversies. Semin Respir Crit Care Med. 2015 Apr;36(2):299-320. doi: 10.1055/s-0035-1547347. Epub 2015 Mar 31.
About the authorView full bio

Dr Toni Hazell, FRCGP
MBBS, BSc, FRCGP, DFSRH, Dip GU med, DRCOG, DCH (London, UK, 2000)
Dr. Toni Hazell qualified from St. Mary’s Hospital Medical School and did her VTS at Northwick Park Hospital.
About the reviewerView full bio

Dr Hayley Willacy, FRCGP
General Practitioner, Medical Author
MBChB (1992), DRCOG, DFFP, MRCOG (Part 1) MRCGP (2007), DFSRH (2013), MSc - medical education (2020)
Dr Hayley Willacy was an NHS GP working in northwest England, who retired from clinical practice in 2022 after 30 years.
Article history
The information on this page is written and peer reviewed by qualified clinicians.
Article also available in English, German, Spanish, French, Italian, Portuguese, Hindi, Hebrew, Arabic, and Swedish.
Next review due: 12 May 2028
16 May 2023 | Latest version

Ask, share, connect.
Browse discussions, ask questions, and share experiences across hundreds of health topics.

Feeling unwell?
Assess your symptoms online for free
Sign up to the Patient newsletter
Your weekly dose of clear, trustworthy health advice - written to help you feel informed, confident and in control.
By subscribing you accept our Privacy Policy. You can unsubscribe at any time. We never sell your data.
More in children's health
- BCG immunisation
- Bedwetting reward systems
- Colic in babies and infants
- Coughs and colds in children
- Erythema toxicum neonatorum
- Hydrocele in infants
- Klinefelter's syndrome
- Left-side abdominal pain in children
- Necrotising enterocolitis
- Neuroblastoma
- Paediatric vulvovaginitis
- Right lower quadrant pain in children
- Safeguarding children
- Tetanus and the tetanus vaccine
- Toddler's diarrhoea
- Turner syndrome
- Undescended testicles
- Urine infection in children
- Wilson's disease