Sickle cell disease
Sickle cell anaemia
Peer reviewed by Dr Toni Hazell, FRCGPLast updated by Dr Hayley Willacy, FRCGP Last updated 21 Feb 2023
Meets Patient’s editorial guidelines
- DownloadDownload
- Share
- Language
- Discussion
- Audio Version
- Add to preferred sources on Google
In this series:Sickle cell trait
Sickle cell disease (SCD) is a serious, inherited condition affecting the blood and various organs in the body. It affects the red blood cells, causing episodes of 'sickling', which produce episodes of pain and other symptoms. In between episodes of sickling, people with SCD are normally well. Long-term complications can occur. Good treatment, started early in life, can prevent complications. So, early diagnosis and specialist treatment are advised for SCD. Sickle cell trait is not the same as sickle cell disease.
At a glance
Sickle cell disease (SCD) is a genetic condition affecting red blood cells, causing them to become sickle-shaped.
Symptoms, like pain episodes and severe infections, usually appear after three months of age and can vary.
SCD is diagnosed by a blood test, which is offered to pregnant women and all newborn babies in the UK.
There is no cure for SCD, but treatments help manage symptoms and prevent complications.
Lifelong monitoring and treatment by a specialist team are essential.
Seek urgent medical help for sudden, severe symptoms like chest pain, high temperature, or sudden severe anaemia.
Sickle cell disease
Sickle cell disease (SCD) is a serious group of conditions which are inherited (genetic). It affects the red blood cells in the blood. Sickle cell anaemia is the name of a specific form of sickle cell disease in which there are two sickle cell genes (see below).
What is sickle cell disease?
In SCD, the red blood cells have a tendency to go out of shape and become sickle-shaped (like a crescent moon) - instead of their normal disc shape. This can cause various problems - as described later. In between the episodes of illness, people with sickle cell disease feel well.
Normal and sickle-shaped blood cells
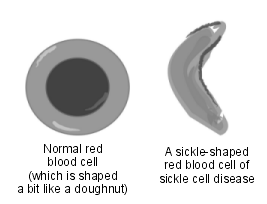
SCD is therefore a group of conditions that cause red cells to become sickle-shaped.
Sickle cell trait is not the same as SCD or sickle cell anaemia. Sickle cell trait means you carry a single sickle cell gene, but it does not normally cause illness.
The rest of this leaflet will discuss SCD, which includes sickle cell anaemia and the other less common disorders.
How common is sickle cell disease?
In the UK, about 12,500 people have SCD. It is more common in people whose family origins are African, African-Caribbean or (less commonly in the UK) Asian or Mediterranean. It is rare in people of North European origin. On average, 1 in 2,400 babies born in England have SCD, but rates are much higher in some urban areas - about 1 in 300 in some places.
SCD is now one of the most common inherited conditions in babies born in the UK.
What causes sickle cell disease?
The cause is inherited (genetic). It is a change in the genes which tell the body how to make an important protein called haemoglobin. To get SCD, you need to have two altered haemoglobin genes, one from each parent. If you only have one of these genes, you will have sickle cell trait, which is very much milder.
Sickle cell inheritance
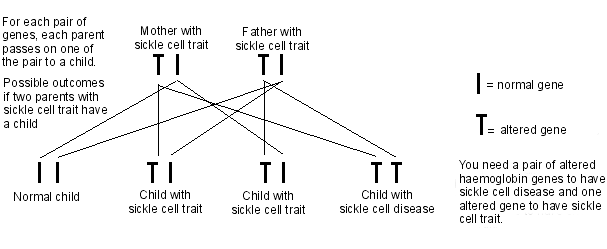
The most common type of sickle cell disease occurs where you have two sickle cell genes (sickle cell anaemia). The medical shorthand for this is haemoglobin SS (or HbSS). Other types of SCD involve one sickle cell gene plus another abnormal haemoglobin gene of a different type. These include: haemoglobin SC; haemoglobin S/beta thalassaemia; haemoglobin S/Lepore; haemoglobin SO Arab.
The symptoms, diagnosis and treatment are similar for all the sickle cell conditions.
How is sickle cell disease passed on?
Sickle cell genes affect the production of an important chemical called haemoglobin. Haemoglobin is located in red blood cells, which are part of the blood. Haemoglobin carries oxygen and gives blood its red colour.
The sickle cell genes make the body produce abnormal haemoglobin called HbS. (Normal haemoglobin is called HbA.) HbS behaves differently from HbA. Under certain conditions, HbS makes the red blood cells change shape - instead of the normal doughnut shape, they become sickle-shaped, like a crescent moon. This is called sickling. Conditions which trigger sickling are cold, infection, lack of fluid in the body (dehydration), low oxygen, and acid (acid is produced in hard physical exercise).
What happens to the sickle cells?
The sickle cells containing mostly HbS are harder and less flexible than normal red blood cells. So, they can get stuck in small blood vessels and block them. This can happen quite suddenly, causing various symptoms which are known as a sickle cell crisis (explained below). Repeated blockages can also lead to complications occurring.
The sickle cells are destroyed more easily than normal red blood cells. This means that people with SCD tend to have a lower number of red blood cells and have a moderate and persistent anaemia. A moderate anaemia is not usually a problem because the HbS (the different haemoglobin) carries oxygen well, and the body can compensate. However, you may get bouts of severe anaemia for various reasons. For example, if too much blood goes to the spleen, if too many red blood cells break down at the same time, or due to certain infections which stop blood cells being made. A severe anaemia can make you very ill.
How is sickle cell disease diagnosed?
The diagnosis is made by a blood test. The blood sample is analysed to see what type of haemoglobin is present in the blood (using a test called haemoglobin electrophoresis or other methods). This can diagnose most cases of sickle cell trait and sickle cell disease (SCD). Sometimes, the result is unclear and extra tests such as DNA (genetic) tests are needed.
The diagnosis is made by a blood test. The blood sample is analysed to see what type of haemoglobin is present in the blood.
Policies for screening for sickle cell disease for pregnant women and babies vary throughout the UK. The policies vary dependent on whether you live in an area where more or fewer than more than 1.5 babies out of every 10,000 births is born with sickle cell disease.
Tests for pregnant women
If the result shows that you carry a sickle cell gene then a test is also offered to the baby's father (if possible). The results of both parents' tests will help to decide whether there is a risk of having a child who is affected by SCD. The results will be explained to you.
If there is a chance that the baby could inherit SCD, you will be offered counselling to discuss whether you would like to have a further test for the unborn baby - a prenatal test. A prenatal test finds out whether the unborn baby actually has SCD. If so, you will be offered counselling to discuss how this could affect the baby and whether you want to continue with the pregnancy.
Would SCD make my baby ill during pregnancy?
No; it does not affect the baby while in the womb. Signs and symptoms of sickle cell disease start from around age 3 months, and treatment for SCD should begin by this age.
When is the best time to have a test?
If you are pregnant, the ideal time to have a sickle cell blood test for yourself is before you are 10 weeks pregnant. This allows more time to test your partner or your baby, if needed. You can ask your doctor for a test early in pregnancy if it is not already offered to you at that time. However, tests can still be done at a later stage.
A prenatal test (on the unborn baby) can be done from 10 weeks of pregnancy onwards, depending on the type of test used. The usual tests offered are chorionic villus sampling (CVS) or amniocentesis.
Should I and my partner have tests before starting a family?
Women or couples may want to be tested for sickle cell trait before starting a family, especially if their family origins make sickle cell trait more likely. The UK's Sickle Cell Society and many health professionals encourage awareness of sickle cell trait and early testing. You can ask your doctor for a sickle cell blood test.
The advantage of having tests before you become pregnant is that you will know whether or not there is a possibility that your baby could inherit SCD. This may be helpful when making decisions about pregnancy. For example, you may want to have a prenatal test during pregnancy if there is a risk of SCD for the baby.
The current recommendations are that women who are undergoing tests for infertility and women who are receiving infertility treatment should be tested for sickle cell trait.
Tests for newborn babies
In the UK, all newborn babies are offered a bloodspot test at 5-8 days after birth. This tests for a number of medical conditions which are considered important because early treatment makes a difference. The test is done by taking a small spot of blood from the baby's heel. The bloodspot test now includes testing for SCD throughout the UK. You will be given the results about six weeks later.
If the baby has sickle cell trait, no action or treatment is needed. If the baby has SCD, the result will be explained. You will be given a clinic appointment to check the diagnosis and to start treatment. Treatment should begin by the time the baby is 3 months old.
Sickle cell disease symptoms
For those living wth sickle cell disease, symptoms of SCD come and go. Usually there are bouts (episodes) of symptoms but, in between episodes, you feel well. The reason that symptoms come and go is that the red blood cells can behave normally for much of the time - but if something makes too many of them sickle, the sickle cells cause symptoms. If there are severe and sudden symptoms due to sickling, this is called a sickle cell crisis.
There is a lot of individual variation in symptoms - how many and how often you get them. Some people with SCD have frequent symptoms, while others have very few and their SCD is hardly noticeable. For most people, symptoms are somewhere in between these two extremes. Most people with SCD have a few episodes of sickle cell crisis each year.
Symptoms usually begin after a few months of age. (Before that age, the baby has a different haemoglobin, called fetal haemoglobin, which is not affected by the sickle cell gene.)
The various symptoms that can occur if you have SCD include:
Episodes of pain
These are also called a pain crisis or a vaso-occlusive crisis. They occur when sickle cells block small blood vessels in bones, which causes pain. Pain usually occurs in bones and joints. The pain can vary from mild to severe, and may come on suddenly.
A common symptom in babies and young children is small bones in the fingers and toes becoming swollen and painful - this is known as dactylitis.
Episodes of tummy (abdominal) pain can occur if sickle cells block blood vessels in your abdomen.
Acute chest syndrome
This occurs when there are blocked blood vessels in the lungs and can sometimes occur with a lung infection.
The symptoms can include chest pain, high temperature (fever) and shortness of breath. Babies and young children may have more vague symptoms and look generally unwell, be lacking in energy (lethargic), be restless or have fast breathing. Acute chest syndrome is very serious and, if it is suspected, you should be treated urgently in hospital.
Acute chest syndrome can start a few days after a painful episode of sickle crisis. It is most common in women who are pregnant or who have recently had a baby.
Infections
People with SCD are more prone to severe infections, particularly from certain types of germs (bacteria), which can cause pneumonia, meningitis, septicaemia or bone infections. (These include the pneumococcal, Haemophilus influenzae type b and meningococcal bacteria, and salmonella bacteria which can infect bones.) Symptoms of infection include fever, feeling generally ill, and pain in the affected part of the body.
Children with sickle cell disease have a high risk of getting severe or life-threatening infections. It is important to see a doctor quickly if you suspect an infection or feel unwell.
Note: a fever can occur in a sickle cell crisis without having an infection.
Sickle cell anaemia
Anaemia is a lack of haemoglobin in the blood. As mentioned above, people with SCD will usually have a moderate anaemia, which does not usually cause problems. However, at times, people with SCD can get a severe anaemia, which can be serious. It may come on very suddenly or more gradually. Urgent treatment may be needed.
Symptoms of severe anaemia are:
Feeling tired, faint, short of breath, dizziness, feeling sick (nausea) or having fast breathing - worse with physical activity.
Babies and small children may be lethargic, not feeding much or generally unwell.
A pale skin colour (easiest to see in the lips, tongue, fingernails or eyelids).
With children, the spleen sometimes enlarges quickly and causes sudden severe anaemia. The enlarged spleen is in the abdomen and can be felt. Parents may be shown how to feel their child's spleen. If the spleen enlarges quickly, it is a sign that urgent treatment is needed.
How can sickle cell disease be treated?
As a rule, there is not a cure for sickle cell disease, so lifelong treatment and monitoring are needed. There are a number of different treatments which help to prevent sickling episodes, or prevent related problems such as infection.
Principles of treatment
You should be treated by a specialist doctor or team, experienced in treating patients with SCD. If the specialist is a long way from your home then some of your treatment may be with a more local hospital or doctor - but the local doctors should get advice from your specialist.
Because symptoms of SCD can start suddenly, you should be able to see a doctor and get hospital treatment urgently, as and when needed.
You can be shown how to recognise symptoms (in yourself or your child), so that treatment can be started quickly.
Treatment should be tailored to your individual needs. New treatments are always being developed, such as crizanlizumab which reduces the number of crises.
It is important to take preventative treatments against infection and to attend your check-ups.
Stem cell transplant is the only available treatment that can cure SCD. It is only used for severe SCD. Its use is limited by side-effects of the procedure and the availability of suitable donors.
Staying healthy
A daily antibiotic is usually recommended (penicillin, or erythromycin if you are allergic to penicillin). This is especially important to protect against serious infections in children aged under 5 years.
Immunisations: all the usual childhood vaccinations are advised, PLUS you should have vaccinations against meningitis and hepatitis B, PLUS a flu (influenza) vaccination once a year. These vaccines are recommended both for adults with SCD and for children with SCD.
Vitamin supplements: extra folic acid is usually recommended. This helps the body to make new red blood cells.
Travel: if you go to a country where there is malaria, be extra careful to take malaria prevention medication and to prevent mosquito bites (people with SCD can get very ill from malaria).
Avoid smoking (which is bad for blood vessels) and excess alcohol.
Avoid factors which can trigger sickling
Factors which can trigger sickling include:
Cold.
Lack of oxygen.
Lack of fluid in the body (dehydration).
Hard exercise.
High temperature (fever).
Infection.
So it can help to:
Drink plenty of fluid.
Take regular exercise (but avoid over-exertion) and eat a healthy, balanced diet.
Avoid getting cold; wrap up well. Avoid over-exertion.
Treat infections and fevers quickly. You will usually be given detailed advice about how to check for signs of fever or infection in yourself or your child, and how to get treatment quickly.
See a doctor quickly if you feel unwell. Tell doctors and nurses that you have SCD.
Treatment of sickling episodes
The vast majority of people who have a sickle cell crisis do not need to be admitted to hospital for treatment. If the pain is mild and there is no fever then it can be possible to be treated at home. Treatment usually involves:
Painkillers
Depending on the amount of pain, you can take various types of pain medication. Mild painkillers are paracetamol or ibuprofen. Moderate ones are codeine or dihydrocodeine. A strong painkiller such as morphine may be needed for severe pain - this is usually given in hospital.
Good hydration
This usually means drinking extra fluid, or sometimes a drip into one of your veins, which is needed if you are more unwell or cannot drink.
Oxygen
This is usually given to you through a face mask in hospital. If you are not getting enough oxygen then more of your red cells may become sickle-shaped.
Antibiotics
These are used if you have an infection, or when infection is suspected. (You will normally be taking a regular preventative antibiotic already, as explained above. However, if an active infection is suspected, you will need a different antibiotic in a higher dose.)
Avoid potential triggers
People with SCD should try to avoid any potential triggers for a sickle cell crisis as much a possible. For example, try to keep warm in cold weather, try to avoid becoming dehydrated and take precautions if you undergo extreme exercise.
Blood transfusions
Blood transfusion is a useful treatment for some situations, such as acute chest syndrome or severe anaemia. It can also be used to help prevent or treat certain complications. The transfusion helps because it adds normal red blood cells to the blood. This corrects anaemia and reduces the sickle-shaped red blood cells. There are potential side-effects from blood transfusions such as iron overload and problems with the immune system. Therefore, transfusions are given for a specific need, rather than routinely.
Treatment of acute chest syndrome
For acute chest syndrome, some of the treatment is the same as for sickling episodes (above) - painkillers, hydration and antibiotics. Also, you may need a blood transfusion and oxygen. A type of chest physiotherapy called incentive spirometry also helps.
Hydroxycarbamide
Hydroxycarbamide (also called hydroxyurea), taken regularly, may help to reduce the amount of symptoms such as pain episodes and acute chest syndrome. Hydroxycarbamide can have serious side-effects and needs monitoring with blood tests. It may be an option but you and your doctor need to think about the pros and cons of taking it.
Women's health
Contraception.
The choice of contraceptive method needs to be considered carefully. The intrauterine contraceptive device (sometimes called 'the coil') may cause particularly heavy painful periods. The use of injectable contraceptives (such as Depo-Provera®) has been reported to provide some protection against sickling episodes.
Planning a baby and pregnancy.
Having SCD increases the risk of certain problems in pregnancy, such as high blood pressure or premature birth. Also, your SCD symptoms might increase while you are pregnant. Be aware that some medications such as hydroxycarbamide should be avoided if you are trying to conceive or become pregnant. You will also be advised to take a higher dose of folic acid (5 mg) if you are pregnant or planning to become pregnant. So, when planning a pregnancy or when pregnant, see your doctor early on. You will normally have extra monitoring from a specialist during your pregnancy.
You may wish to have tests for your partner and unborn baby, to find out whether your baby could inherit SCD.
Anaesthetics and operations
An operation or anaesthetic is one of the things that can trigger sickling. Therefore, always tell your anaesthetist, surgeon and other healthcare staff that you have SCD, so that precautions can be taken to reduce the risk of sickling. For example, sometimes a blood transfusion before the operation or anaesthetic may be advised.
What are the complications of sickle cell disease?
Possible complications in children
Growth, development and nutrition
As with any long-term illness, a child with SCD may grow more slowly than usual, or be undernourished if the illness affects their appetite. Your child's growth, development and nutrition should be checked regularly, and nutritional supplements may be given if needed.
Some children with SCD take longer than usual to gain control of their bladder at night, so may wet the bed (nocturnal enuresis). Various treatments can help.
For teenagers, puberty may start about 2-3 years later than average.
The growth of bones can also be affected. For example, there may be changes in the hip or shoulder joints due to blocked blood vessels in that part of the bone. If a joint is severely affected, surgery may be needed.
Stroke or brain injury
This is a serious complication of sickle cell disease and affects about 1 in 10 children or teenagers with SCD. If sickle cells block blood vessels in the brain, this may cause a stroke. There may be symptoms of stroke such as weakness of the face or limb, or speech difficulty. For some children, there may be no obvious symptoms. However, many tiny strokes may cause a subtle brain injury and make learning more difficult.
Strokes are treated with blood transfusion, which improves blood flow to the brain. Also, research has found that regular blood transfusions help to prevent strokes. An ultrasound test called a transcranial Doppler can be used to look at the blood flow to the brain. This helps doctors to decide whether your child needs blood transfusions for prevention. Children aged 3 years should be offered these scans.
Spleen problems
The spleen is an organ located in the tummy (abdomen), in the top left-hand side. Its function is to help the immune system. Sickle cells can block blood vessels in the spleen. This can make the spleen swell up suddenly with blood - in effect, it is like losing blood into the spleen. This is one cause of sudden and severe anaemia, when your child becomes suddenly ill. The medical term is splenic sequestration. It needs urgent treatment with a blood transfusion.
If this problem happens more than once then one option is surgery to remove the spleen. However, by adulthood the problem normally resolves because the spleen becomes hard (fibrosed) and cannot swell.
Parvovirus infection
Parvovirus is a common infection in childhood. Normally it causes a mild illness with high temperature (fever), flushed cheeks and a rash. With SCD, the virus can upset the bone marrow, which then stops making blood for a while. This causes a severe anaemia and needs treating with blood transfusions until the bone marrow recovers.
Complications of blood transfusions
Transfusions can cause blood reactions. These are less likely if the blood is carefully matched to your blood type. Infections such as hepatitis B and C can be transmitted by transfusion. This is less likely in the UK and countries where donor blood is tested for infections. Hepatitis B vaccination is also recommended.
Repeated blood transfusions can overload the body tissues with iron. You may need tests to measure the iron level in the body. If iron levels get high, you may need treatment called chelation, which helps the body get rid of excess iron.
Possible complications in older teenagers and adults
Organ damage can develop gradually during teenage and adult years, due to repeated, small blockages of tiny blood vessels. The amount of complications can vary from person to person.
Lungs, heart and kidneys
Any of these organs may suffer some damage. Therefore, you will normally be offered regular checks on your heart, lungs and kidney function. Various treatments can help.
Eyes
Regular eye checks are important. SCD may cause changes to blood vessels in the back of the eye (retina); this is called retinopathy. For retinopathy, laser treatment is given to prevent further damage.
Also, sickle cells may cause sudden blockage of a blood vessel in the eye. If this happens, you will have a sudden reduction in your vision. This needs immediate treatment. So, always see a doctor quickly if your vision reduces suddenly in any way.
Unwanted erections
Some teenage boys and men with SCD may get unwanted erections of the penis, which may be painful. The medical name for this is priapism. This can be quite brief but if an erection does not subside within one hour then urgent treatment is needed. There are various treatments to relieve or prevent unwanted erections.
Gallstones
Stones in the gallbladder are more common in people with SCD, and can cause bouts of pain in the upper right side of the abdomen. They may need treatment which is usually an operation to remove the gallbladder.
Leg ulcers
Leg ulcers can occur with SCD but are not common. Treatment is with dressings, and zinc supplements may help.
Complications of blood transfusions
These are explained above for children, and also apply to adults.
What is the outlook?
Sickle cell disease (SCD) is a serious condition which may shorten life. Without treatment, people with SCD may die in childhood, from problems such as infection. Good treatment makes a great difference. Improvements in treatment mean that life expectancy has increased.
Even with modern treatment, SCD can still cause serious or life-threatening problems. Dangerous problems are severe infection, acute chest syndrome and sudden severe anaemia. Awareness of symptoms and early treatment are important.
There is a lot of individual variation in the severity and outlook (prognosis) for SCD. Some people get very few problems from their SCD; others have more symptoms or more complications.
The treatment of sickle cell anaemia is a developing area of medicine. New treatments continue to be developed and the information on outlook above is very general. The specialist who knows your case can give more accurate information about the outlook for your particular situation.
Patient picks for Anaemia

Allergies, blood and immune system
Folic acid deficiency
A normal balanced diet contains enough folic acid. However, a folic acid deficiency may cause anaemia and sometimes other symptoms.
by Dr Rosalyn Adleman, MRCGP

Allergies, blood and immune system
Vitamin B12 deficiency and pernicious anaemia
Vitamin B12 is essential for life. It is needed to make new cells in the body, such as the many new red blood cells which are made every day. Vitamin B12 is found in meat, fish, eggs and milk - but not in fruit or vegetables. A normal balanced diet must contain enough vitamin B12. A lack of vitamin B12 leads to anaemia and sometimes to other problems.
by Dr Colin Tidy, MRCGP
Frequently asked questions
What is the difference between sickle cell disease and sickle cell trait?
Sickle cell disease (SCD) is a group of serious inherited conditions where red blood cells become sickle-shaped, causing various health problems. Sickle cell anaemia is a specific type of SCD where an individual has two sickle cell genes. In contrast, sickle cell trait means a person carries only one sickle cell gene and typically does not experience illness or symptoms.
Why do symptoms of sickle cell disease often start around 3 months of age?
Symptoms of sickle cell disease usually begin after a few months of age because babies are born with a different type of haemoglobin, called fetal haemoglobin, which is not affected by the sickle cell gene. This fetal haemoglobin protects them initially, but as it is replaced by adult haemoglobin, the effects of the sickle cell gene become apparent.
Are there any specific considerations for women with sickle cell disease regarding contraception?
Yes, for women with sickle cell disease, the choice of contraception needs careful consideration. An intrauterine contraceptive device (coil) might lead to particularly heavy and painful periods. On the other hand, injectable contraceptives (like Depo-Provera®) have been shown to offer some protection against sickling episodes.
Why might children with sickle cell disease have delayed puberty?
As with many long-term illnesses, children with sickle cell disease may experience growth and developmental delays. This can include puberty starting later than average, typically about 2-3 years after their peers. Regular checks on growth, development, and nutrition are important.
How does hydroxycarbamide (hydroxyurea) help people with sickle cell disease?
Hydroxycarbamide, when taken regularly, can help reduce the frequency of symptoms such as pain episodes and acute chest syndrome in people with sickle cell disease. However, it can have serious side-effects and requires careful monitoring with blood tests. The decision to use it involves weighing the potential benefits against these risks.
What specifically causes pain during a sickle cell crisis?
Pain during a sickle cell crisis, also known as a vaso-occlusive crisis, occurs when the sickle-shaped red blood cells block small blood vessels, particularly in the bones. This blockage restricts blood flow and oxygen to the affected areas, leading to pain that can range from mild to severe and may come on suddenly.
Further reading and references
- Management of Sickle Cell Disease in Pregnancy; Royal College of Obstetricians and Gynaecologists (August 2011)
- Sickle cell acute painful episode; NICE Clinical Guideline (June 2012)
- Guidelines on red cell transfusion in sickle cell disease Part II - indications for transfusion; British Committee for Standards in Haematology (2016)
- PHE; Sickle cell and thalassaemia screening: programme overview, 2013.
- Crizanlizumab for preventing sickle cell crises in sickle cell disease; NICE Technology appraisal guidance, November 2021
- Oteng-Ntim E, Pavord S, Howard R, et al; Management of sickle cell disease in pregnancy. A British Society for Haematology Guideline. Br J Haematol. 2021 Sep;194(6):980-995. doi: 10.1111/bjh.17671. Epub 2021 Aug 19.
- Sickle cell disease; NICE CKS, July 2021 (UK access only)
About the authorView full bio

Dr Hayley Willacy, FRCGP
General Practitioner, Medical Author
MBChB (1992), DRCOG, DFFP, MRCOG (Part 1) MRCGP (2007), DFSRH (2013), MSc - medical education (2020)
Dr Hayley Willacy was an NHS GP working in northwest England, who retired from clinical practice in 2022 after 30 years.
About the reviewerView full bio

Dr Toni Hazell, FRCGP
MBBS, BSc, FRCGP, DFSRH, Dip GU med, DRCOG, DCH (London, UK, 2000)
Dr. Toni Hazell qualified from St. Mary’s Hospital Medical School and did her VTS at Northwick Park Hospital.
Article history
The information on this page is written and peer reviewed by qualified clinicians.
Article also available in English, German, Spanish, French, Italian, Portuguese, Hindi, Hebrew, Arabic, and Swedish.
Next review due: 20 Feb 2028
21 Feb 2023 | Latest version

Ask, share, connect.
Browse discussions, ask questions, and share experiences across hundreds of health topics.

Feeling unwell?
Assess your symptoms online for free
Sign up to the Patient newsletter
Your weekly dose of clear, trustworthy health advice - written to help you feel informed, confident and in control.
By subscribing you accept our Privacy Policy. You can unsubscribe at any time. We never sell your data.